Risk Management in Single-Use Technology

Risk management is pervasive throughout the biopharmaceutical industry. It is an important factor during the implementation of new equipment or procedures into an operation. Likewise, risk management is also key when assessing the impact of changes. When doing a root cause analysis, evaluation of the risks becomes central to define the potential solutions to the problem analyzed and to eliminate the contributing cause.
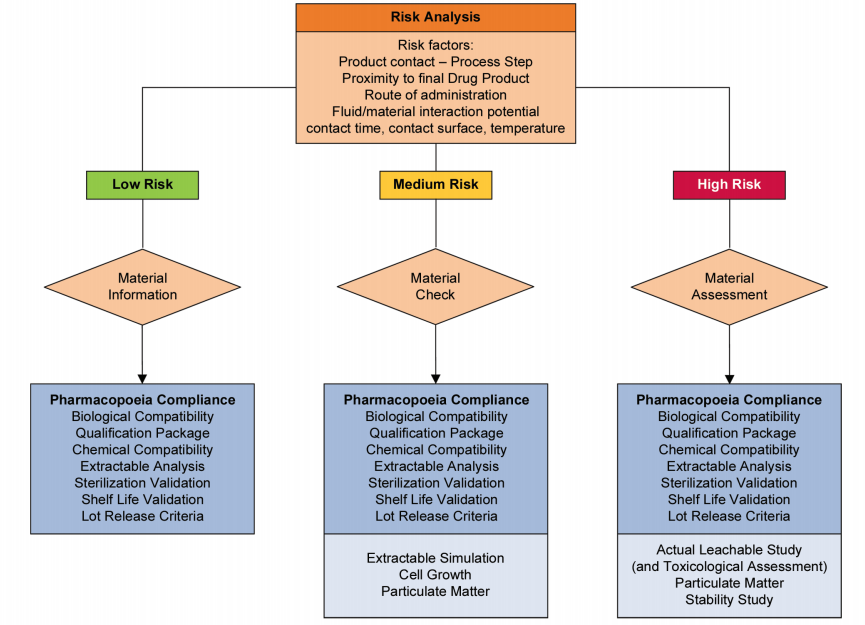
Risk management should be applied throughout the implementation of single-use technology. The guidance from ASTM E2500-13 details the role and interrelationships of risk management as part of Good Engineering Practice.2 (p. 4, Figure 1) Risk assessments should be conducted at the beginning and multiple subsequent points of the implementation program. To be effective, risk management programs require involvement from top management and support across the entire organization.
One of the guiding principles of ICH-Q9 is: “the level of effort, formality, and documentation of the quality risk management process should be commensurate with the level of risk”.3 The ISPE Good Practice Guide: Good Engineering Practice further encourages biopharmaceutical companies to apply the appropriate level of risk management to provide cost-effective solutions. That guide classifies risk management as a core activity that should balance risk against benefits and that risks should be reduced to acceptable levels.4
It is recommended that the risk management program include, at a minimum, a failure mode and effects analysis (FMEA) done at the beginning of the implementation to address at least the potential risks. Refer to the ISPE Baseline® Guide: Commissioning and Qualification5 and ASTM E2500-132 . Table 2 identifies notable tools and resources to assess risk.
Categories of Risks
The sources of risks identified earlier in this article can be addressed in a comprehensive risk analysis. Clarification of each type of risk can help to identify and structure the criteria to include in the risk assessment.
Risks due to leaks or performance issues usually tend to surface once the single-use product is applied in manufacturing operations. Because many connections occur in single-use assemblies and a significant amount of handling can occur during product installation and use, it is important to estimate this risk early. Historical information from the supplier relative to failure rates and trends will help assess this risk.
Experience with the single-use product during test programs can also be used to assess the risk if the test conditions were conducted in a controlled manner. Evaluation of single-use products during test programs executed by the user can present variable and unusual conditions to the single-use products as the application (circumstances of use) is developed. Therefore, leaks and performance issues that occur in this phase should be adjusted for the more controlled conditions that are present in a manufacturing cGMP environment. Leaks or performance issues discovered in the user’s test programs should be used to refine manufacturing standard operating procedures (SOPs) or to initiate design changes in single-use assembly and/or shipping containers that minimize this risk. The level of adherence to refined SOPs in manufacturing environments will typically align with the predictability of risk for leaks or performance factors.
| Change Category | Examples of Changes |
|---|---|
| Maintenance/improvement changes |
Operational improvements Change in drawing format or documentation Different storage location Change in transit route |
| Major disruptive changes | Change in material of construction or formulation New manufacturing location Change in component dimensions Change in packaging of single-use component/assembly Different single-use product supplier New sterilization supplier/location |
| FMEA |
| Fault tree analysis |
| ICH Q93 |
| ASTM E2500-132 |
| ISPE Guide: Science and Risk-Based Approach for the Delivery of Facilities, Systems, and Equipment6 |
| ISPE Baseline® Guide: Commissioning and Qualification5 |
Risks due to availability of single-use products to meet production schedules depend primarily on the reliability of the supplier. These risks also must be minimized for smooth operations. Keeping a high level of inventory can offset some availability risks. However, keeping a high inventory is costly, and this approach can cause large inventory fluctuations when inventory needs to be discarded due to shelf-life constraints.
The physical size of the single-use assembly can be a risk factor to consider. The larger assemblies have characteristics that can increase risk. This include:
- They have higher surface contact area.
- They contain larger volumes of liquid.
- They have more connections.
- They are more complex.
- They are heavier.
These factors can make it more difficult for the operators to handle the assemblies from the package to their installation into the process/operation.
Although all risk factors need to be evaluated in the assessment, one of the more important risk categories for single-use technology involves materials. This is because the single-use components and assemblies are often customized and implemented in new applications. Therefore, the rest of this article focuses on a consistent compliance approach to demonstrating suitability of a given single-use system (SUS) for its intended use in manufacturing. Different applications of SUT are guided by user requirements, which guide the total requirements for design, selection, qualification, procurement, and implementation considerations.
Risk Assessment Approach
The risk assessment model presented in the ISPE Good Practice Guide: Single-Use Technology involves the calculation of a risk score representing the potential risk of the process contact materials (PCM) that are present in the SUS1 . For a given risk category, additional required in-house qualification should be performed, or leveraged from existing data, and documented. Supplier and, if necessary, in-house qualification data packages are required to demonstrate suitability of the process contact materials for its intended use. Where a prior history of using single-use technology has been documented, a comparability protocol can be considered in addition to leveraging the existing data for qualification attributes.
The steps for the risk assessment model are:
- Step 1: Identify user requirements for the process contact materials.
- Step 2: Obtain process contact materials validation data package from the supplier.
- Step 3: Perform risk assessment.
- Step 4: Execute in-house qualification studies required based on the risk score.
- Step 5: Perform risk mitigation.
The steps to qualifying process contact materials for manufacturing applications include identifying specific supplier qualification requirements, which are drawn from the following 15 critical qualification attributes (full descriptions of these 15 attributes are provided in Appendix 4 of the ISPE Good Practice Guide: Single-Use Technology;1 other qualification attributes specific to an application may be added):
- Biocompatibility testing
- Mechanical properties
- Gas transmission properties
- Gompendial physicochemical testing
- Animal origin control
- Total organic carbon analysis
- pH and conductivity
- Extractables and leachables
- Chemical compatibility
- Protein adsorption studies
- Endotoxin testing
- Sterilization (irradiation)
- Container closure integrity
- Particulate testing
- Calibration of embedded instrumentation
Some of these attributes, such as animal origin control, may become core requirements for a firm. Others, such as calibration of embedded instrumentation for bioreactors, may be added depending on the application.
| Risk Classification | Route of Administration | Examples of Drug Formulation |
|---|---|---|
| High (risk score = 10) | Inhalation/nasal | Inhalation aerosols and solutions Nasal sprays Nasal aerosols Inhalation powders |
| Injection (>10 exposures per life) | Injectable suspensions and solutions Sterile powders; powders for injection |
|
| Ophthalmic (>10 exposures per life) | Ophthalmic solutions and suspensions | |
| Medium (risk score = 5) | Injection (≥ 10 exposures per life) | Injectable suspensions and solutions (e.g., vaccines) Sterile powders; powders for injection |
|
Ophthalmic (≥10 exposures per life) |
Ophthalmic solutions and suspensions | |
| Internal application | Implants Rectal/vaginal creams and solutions |
|
| Low (risk score = 1) | Transdermal | Transdermal ointments, creams, lotions, and patches |
| Internal irrigation | Nasal rinse solutions | |
| Topical | Topical lotions, creams, solutions, and suspensions Topical powders Topical aerosols |
|
| Oral | Lingual aerosols Oral solutions and suspensions Oral powders Oral tablets Oral capsules (hard and soft gelatin) |
In advance of conducting qualification activities, it is recommended that an audit of the supplier be completed. This audit can determine in advance how well the process contact materials might perform against applicable elements for qualification. Once the results of the risk assessment are known and the elements required to be taken into consideration for qualification have been identified, process contact materials from appropriate suppliers are chosen for qualification activities.
If qualification results are inconclusive or variable between process contact materials batches, additional controls may need to be placed on the process contact materials as part of the supplier manufacturing process or when process contact materials are received by the end user for manufacturing. The qualification results ultimately lead to the incoming requirements as part of the normal quality acceptance of process contact material batches.
Other requirements that should be taken into consideration include:
- Establish the expiration dates for the process contact material, with sufficient justification and supporting documentation.
- Provide storage conditions requirements necessary to support the expiration date.
- Determine the applicability of any preuse tests.
- In addition to any incoming process contact material testing, confirmation of the quality of the process contact material may be needed when it is installed or just prior to use.
- In the absence of specific guidelines, an evaluation and justification should be made to establish any requirements.
- Determine the need of any postuse tests.
- There may be instances where there is a regulatory requirement to confirm integrity postuse (e.g., vent filters).
- In the absence of specific guidelines, an evaluation and justification should be made to establish any requirements.
Risk Assessment—Calculation of Risk Score
The purpose of the risk assessment is to determine the extent of in-house qualification required according to a calculated risk score for each process contact material. The risk assessment model presented here takes into account the potential risk to product quality and patient safety. Certain risks should be mitigated by supplier quality systems and upfront evaluation such as chemical compatibility and USP Class VI certification.7 Supplier audits should be performed to ensure full traceability of the process contact material to its raw materials.
In advance of conducting qualification activities, it is recommended that an audit of the supplier be completed. This audit can determine in advance how well the process contact materials (PCM) might perform against applicable elements for qualification.
As an example, a risk assessment model may be formulated to calculate the risk score as follows:
Risk Score = A × B × C × D
Where:
A = Route of administration
B = Proximity to final product
C = Contact time
D = Surface area–to-volume ratio
This risk assessment model can be applied to assess the relative risk of an individual process contact material and to determine the amount of in-house qualification data required. For each risk factor, the score can be classified as high (risk score = 10), medium (risk score = 5), or low (risk score = 1).
Risk Factor A: Route of Administration
For the risk assessment, the following information relevant to the route of administration should be recorded:
- Product name (that the material will be used with)
- Statement of the dosage form
Table 3 lists examples of risk score assignments for routes of administration along with selected examples of drug formulation. The risk classification for the route of administration is based on the US Food and Drug Administration publication Guidance for Industry: Container Closure Systems for Packaging Human Drugs and Biologics—Chemistry, Manufacturing, and Controls Documentation.8
Risk Factor B: Proximity to Final Drug Product
The likelihood that the process contact material will have an impact on the quality of the drug product generally increases as the process moves downstream toward the manufacture of final drug product. In certain cases, such as biologics, the risk may also be high at vulnerable upstream points. Knowledge of the process, the contact materials, and the product should be applied to assign an appropriate risk level. It is therefore important for the validation team to collaborate with the formulation team to understand these sensitivities and requirements. Table 4 lists examples of risk score assignments based on proximity to final product.
Risk Factor C: Contact Time
Contact time is the total exposure time that the process contact material is in contact with the product (solution). If the solution just flows through, then the contact time is short and can be rated as low risk. Contact times would likely be long during the down/dwell time when the solution is in static contact with the process contact material, or during the entire mixing period where solutions are agitated in the mixing tank/bag. If the solution is flushed after the stoppage, the static time during the stoppage is the largest contributor to the contact time and can often be used to determine the risk score for contact time.
If the product is in solid phase, it should be rated as low risk regardless of the contact time. Table 5 lists examples of risk score assignments based on contact time.
Risk Factor D: Surface Area–to–Volume Ratio
The greater the ratio of the contact material surface area to the product volume (such as batch size) is, the greater the potential risk for leachables, adsorption or absorption of active ingredients or excipients, and chemical reactions with the contact material.
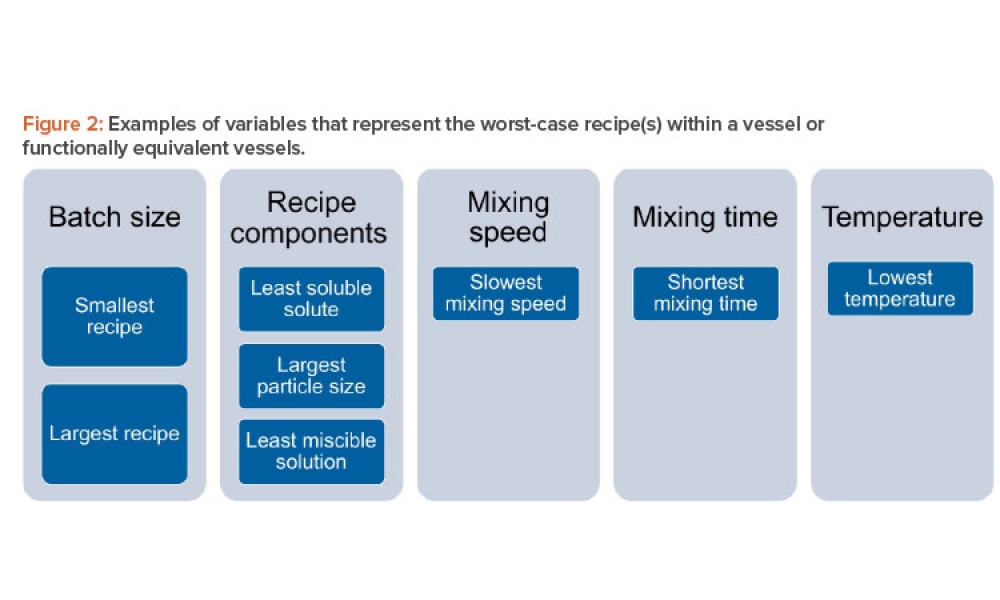
The worst-case surface area-to-volume ratio is a single-use product/assembly with a smaller process volume because it usually has higher surface ar-ea-to-volume ratio. The smallest batch size usually represents the worst-case scenario. The lower the volume is, the more concentrated any potential leachables would be. If the single-use product/assembly applied in the manufacturing process is large (e.g., an assembly containing ≥100 liters of fluid), using a very small amount of single-use material for testing (e.g., a 50-mL container) may not provide data usable for interpretation. A higher amount of single-use material (e.g., a 150-mL, 250-mL, or even 500-mL container) will provide better data for extrapolation to manufacturing-size single-use products/assemblies.
The likelihood that theprocess contact material will have an impact on the quality of the drug product generally increases as the process moves downstream toward the manufacture of final drug product. In certain cases, such as biologics, the risk may also be high at vulnerable upstream points.
| Risk Classification for Risk Factor B | Proximity to Final Product | Comment/Justification |
|---|---|---|
| High (risk score = 10) | Manufacture of the dosage form without a dilution or purification step before the final container closure system. |
Any contaminants will be transferred into the container and consumed by patients. |
| Medium (risk score = 5) | Compounding of the drug product that involves a dilution or purification step before filling. Production of active substances that will be >50% concentration in the final drug product. |
All steps including diafiltration, purification, filtration, and/or dilution >50% will provide a synergistic effect in reducing contaminants in the final product. |
| Low (risk score = 1) | Production of active substances including all media and buffer preparation. |
All steps before compounding will inherently have lower risk because all the downstream process steps will reduce/dilute contaminants as the process progress. |
*Not all of the categories for the proximity to final product risk factor listed in this table will be used at every site (e.g., if a site only performs filling of final product, all contact materials will be high risk for that area of production).
| Risk Classification | Contact Time | Comment/ Justification |
|---|---|---|
| High (risk score = 10) | >7 days of exposure time | Single-use technology will be treated as an intermediate/shipping storage vessel if materials will be stored beyond 7 days. |
| Medium (risk score = 5) | Between 48 hours and 7 days of exposure time |
Intermediate or bulk drug product may be stored in bags up to 7 days for further processing. |
| Low (risk score = 1) | <48 hours of exposure time | Production campaigns can be filled within 36 to 48 hours. |
| Risk Classification | Surface Area– to–Volume Ratio |
Comment/ Justification |
|---|---|---|
| High (risk score = 10) | >0.01 cm2/mL | A safety factor of >15-fold relative to extraction condition per USP Class VI testing7 |
| Medium (risk score = 5) | 0.001–0.01 cm2/mL | A safety factor of between 15- and 150-fold relative to extraction condition per USP Class VI testing7 |
| Low (risk score = 1) | <0.001 cm2/mL | A safety factor of >150-fold relative to extraction condition per USP Class VI testing7 |
Surface area can be calculated based on the dimension of the contact materials. For items such as gaskets and O-rings, the surface area in contact with a solution can be estimated. Overestimating the area covers the worst-case scenario.
For gases (e.g., nitrogen), the risk should be classified as low because the risk of a gas removing substances/leachables from the process contact material is very low. Table 6 lists examples of risk score assignments based on the surface area-to-volume ratio.
Determination of Final Risk Level
After the calculation of the final risk score using the risk score equation noted previously (Risk Score = A × B × C × D), the final risk level is assigned as follows:
- Low: calculated risk score ≤1,000
- Medium: calculated risk score between 1,001 and 4,999
- High: calculated risk score ≥5,000
Documentation of the risk score calculation for each process contact material should be included in the process contact material qualification report. The final risk level can be used to determine the additional in-house qualification studies required.
Executing In-House Qualification
Based on the final risk level of the process contact material, the required in-house qualification activities can be determined. Use of subcontractor services for testing may be considered. Regardless of final risk level, in-house qualification requirements should always include:
- Leak/pressure/crack verification
- Tear evaluation (for bags)
- Sterility evaluation (for sterile-supplied process contact material)
- Endotoxin evaluation (for sterile-supplied process contact material)
- Integrity testing (for 0.2-µm filters, whether sterilizing or for bioburden reduction)
Where the final risk level is medium (calculated risk score between 1,001 and 4,999), additional in-house qualification requirements should include:
- Sorption testing (the uptake of product components by the plastic materials)9
- Spallation testing (particle shedding due to repeated compression of peristaltic pump tubing)10
Where the final risk level is high (calculated risk score ≥5,000), additional in-house qualification requirements should include:
- pH change evaluation
- Leachables testing
- Particulate evaluation
Once the required in-house qualification requirements are identified, there are several approaches to reduce the amount of testing, such as using existing data from suppliers and in-house data, the quality-by-design (QbD) approach, paper exercises, or a combination of these methods.
Using Existing Data
In most cases, existing data from the supplier and in-house data can be utilized without the need to perform additional work. Careful evaluation is necessary to ensure that the existing data support the intended use of the process contact material.
Risk is also a consideration in evaluating supplier documentation. Only formal and official documents issued by the supplier should be admissible. Supporting data need to be provided using an appropriate format (e.g., official statement or executive summary) and registered in an adequate tracking system. A traceable formal document from the supplier should be used; use of information from a website is not recommended.
QbD Approach
The QbD approach involves using data for a higher-risk process contact material to qualify the same process contact material in the same or lower-risk categories. The process contact material categorized as high risk can be qualified to bracket lower-risk-category process contact material if all of the following criteria are met:
- Same grade of resin and material of construction
- Same supplier/manufacturer
- Used for the same/similar drug substance, drug product, or excipient
All samples can be prepared in accordance with QbD principles to represent worse-case and bracket uses in less-severe conditions.
Paper Exercise and Combination Approaches
For certain low-risk applications, in-house qualification studies can potentially be satisfied with a paper exercise to meet qualification requirements without the need to perform testing.
A combination of approaches in one process contact material qualification can be considered. If in-house qualification testing is required, the testing can be designed for an individual process contact material or for a group of process contact material. It is important to first understand the advantages and disadvantages of each approach and the future implications with regard to data leveraging, material changes, and alternate supplier qualification.
Risk Mitigation
Following qualification activities, the report conclusion should highlight any of the tests in which additional controls may need to be placed on incoming materials. If the qualification package is complete and compliant, with all acceptance criteria met and without inconclusive test results, the process contact material is routinely accepted with minimal incoming test requirements. These incoming requirements should minimally include:
- Confirmation of materials (correct process contact material and site of manufacture)
- Confirmation of sterility status, if applicable
- Review of the certificate of analysis (COA) for any prescribed supplier testing and certification
- Confirmation of packaging integrity or packaging configuration
If any qualification studies are inconclusive, incoming controls should be placed on the process contact material. Likewise, deviations encountered during the use or processing of the process contact material may also be an indicator for placing controls on the incoming process contact material.
If the material does not meet the minimum requirements, there are remediation options, such as:
- Requesting that the supplier generate the test data
- Performing in-house qualification
- Selecting another supplier
- Implementing a risk-mitigation step in the process, if possible
Risk management involves looking at all the variables and their impact. Effective risk mitigation depends on a thorough assessment of risk for all the materials. Many of the construction materials used for single-use technology may already be in the existing process. The materials may exist in filters, thermoplastic tubing, polycarbonate connectors, silicone tubing, or housings for sensors. Making use of this information can facilitate the risk assessment.
It is also important to understand the context of the risks. For years, the industry has used glass-lined tanks for products/processes sensitive to metal ions or their catalytic effects. An extractables study on type 1 borosilicate glass, typically used for the glass-lined tanks, may identify the presence of lead and arsenic. However, the detection of arsenic and lead in an extraction study does not invalidate the use of the glass-lined tanks. Such an extraction study is considered rather extreme due to its use of nitric acid and a reflux method. Similarly, the extraction methods for polymeric and other single-use materials can be considered extreme, and the results need to be assessed appropriately.
Conclusion
Comprehensive risk management relies on:
- Evaluating risk from all sources
- Including risk of all contact materials (polymeric and other components)
- Assessing risk based on impact
- Balancing risk against benefits
There are numerous publications on handling risks in biomanufacturing, including those referenced in this article. The ISPE Good Practice Guide: Single-Use Technology should be a primary document to use when developing a risk management program. The guide aims to provide direction for managing risk that is practical and effective.

More on Risk in Single-Use Technology
The ISPE Good Practice Guide: Single-Use Technology provides a roadmap for efficient implementation of single-use technology (SUT) with minimum disruptions to existing operations. From this Guide, users will learn:
- How to select single-use components and design functional systems
- When and how to perform effective extractables and leachables studies
- How to evaluate suppliers of single-use technology
- About the interrelated tasks for implementing single-use technology
- 2 a b c American Society for Testing and Materials. ASTM E2500-13, Standard Guide for Specification, Design, and Verification of Pharmaceutical and Bio-pharmaceutical Manufacturing Systems and Equipment. West Conshohocken, PA: ASTM International, 2013.
- 3 a b International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. “ICH-Q9—ICH Harmonized Tripartite Guideline, Quality Risk Management, Q9.” Published 2005. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step4/Q9_Guideline.pdf
- 4ISPE Good Practice Guide: Good Engineering Practice. North Bethesda, MD: International Society for Pharmaceutical Engineering, 2008.
- 5 a b ISPE Baseline Pharmaceutical Engineering Guide, Vol. 5, Commissioning and Qualification. North Bethesda, MD: International Society for Pharmaceu-tical Engineering, 2001.
- 6ISPE Guide: Science and Risk-Based Approach for the Delivery of Facilities, Systems, and Equipment. North Bethesda, MD: International Society for Pharmaceutical Engineering, 2011.
- 1 a b
- 7 a b c d United States Pharmacopeia. “USP<88> In-Vivo Biological Reactivity.”
- 8US Food and Drug Administration. “FDA Guidance for Industry: Container Closure Systems for Packaging Human Drugs and Biologics—Chemistry, Manufacturing, and Controls Documentation.” Published May 1999. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/container-closure-systems-packaging-human-drugs-and-biologic
- 9Bahal, S. M., and J. M. Romansky. “Spalling and Sorption of Tubing for Peristaltic Pumps.” Pharmaceutical Development and Technology 7, no. 3 (2002): 317–23.
- 10Liu, Y., M. Faria, and E. Leonard. “Spallation of Small Particles from Peristaltic Pump Tube Segments.” Artificial Organs 41, no. 7 (2017): 672–7. doi:10.1111/aor.12815