Flexible Facility Design for Multiple Cell Therapy Processes

In many critical ways, the design of facilities for multiple cell therapy processes is unlike the design of conventional pharmaceutical facilities. This article surveys several of the key issues to consider when designing facilities capable of manufacturing multiple cell therapies, including regulatory definitions, product life cycles, processing systems, relevant cell therapy technologies and equipment, biosafety, cross-contamination, facility automation, and layout options. A case study is used to illustrate flexible cell therapy facility design.
The field of cell and gene therapy has come quite a long way since Friedmann and Roblin authored the paper “Gene Therapy for Human Genetic Dis-ease” in 1972.1 The first approved gene therapy clinical research took place in the US under the direction of William French Anderson at the National Institutes of Health (NIH) in 1990,2 and the field has only accelerated since. Through the 2000s and 2010s, numerous advances were achieved in the treatment of cancers and other genetically driven diseases. Most recently, US FDA approvals of cell therapies from Kite Pharma (YESCARTA) and Novartis (KYMRIAH) have shown the power of cell therapies to treat cancers.3 Reading through the history of cell and gene therapy in the context of manufacturing facility design, it becomes increasingly clear how important it is to understand what these therapies are and how they are defined. Common questions for those new to the field are: What are cell therapies? Why do people refer to cell and gene therapies together? The answers are everyone’s least favorite response, “Well, it depends!” In this case, it depends on who you are asking.
The US FDA and the EMA define cell and gene therapies differently. The EMA calls them “advanced therapy medicinal products” (ATMPs),4 and the FDA calls them “human cells, tissues, and cellular and tissue-based products” (HCT/Ps).5 Each regulatory body then provides more precise categorizations (Table 1).
Generally, cell therapies grow cells outside the body and implant whole live cells for a therapeutic benefit. These cells can either be taken from the patient (autologous) or from a donor (allogenic). Cells used for cell therapy are often stem cells (cells that can mature into different types of specialized cells), and they may or may not be genetically altered.
Gene therapies, on the other hand, modify the genes of cells for a therapeutic benefit. This can happen by replacing a disease-causing gene with a healthy copy of the gene, inactivating a disease-causing gene that is not functioning properly, or introducing a new or modified gene into the body to help treat a disease.6 Gene therapy can be either in vivo (inside the patient) or ex vivo (outside the patient).
| Agency | Term | Definition |
|---|---|---|
| US FDA | Gene therapy | Modifies the genes or cells for a therapeutic benefit. |
| Cell therapy | Grows cells outside the body and implants whole live cells for a therapeutic benefit. | |
| Autologous | The source of the cells is the same as the subject to be treated. | |
| Allogenic | The source of the cells is different than the subject to be treated. | |
| EMA | Gene therapy medicines | Contain genes that lead to a therapeutic, prophylactic, or diagnostic effect. They work by inserting recombinant genes into the body. |
| Somatic cell therapy medicines | Contain cells or tissues that have been manipulated to change their biological characteristics or cells. | |
| Tissue-engineered medicines | Contain cells or tissues that have been modified so they can be used to repair, regenerate, or replace human tissue. |
Cell and gene therapy technologies are lumped together because they are linked by commonalities of scientific procedure. For example, several of the leading cell therapies use gene editing methods to make the cells have therapeutic value. In the US, an engineered cell product such as a chimeric antigen receptor T cell (CAR-T) therapy would be called a cell therapy because cells are what is administered to the patient. In the EU, CAR-T therapy is considered a gene therapy because genetic engineering provides the activity to the cells.
Cell Therapy Life Cycle
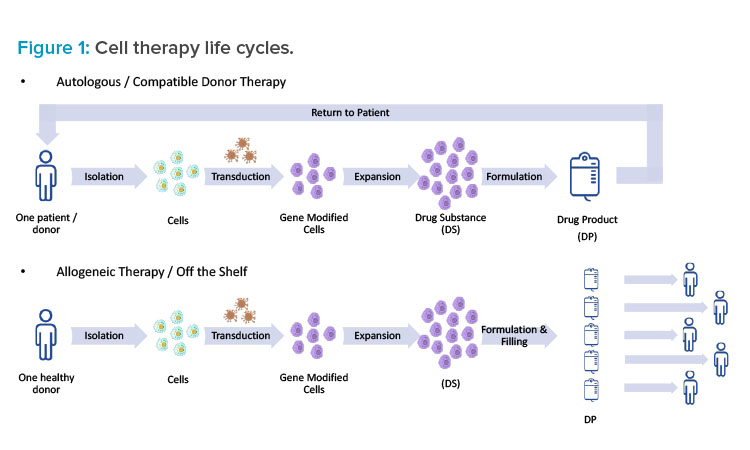
Understanding the life cycle of a specific cell therapy is essential when taking a holistic approach to facility design. In the case of genetically modified autologous therapies, such as CAR-T therapies, there truly is a cycle (see Figure 1). First, cells are isolated from a single patient in a clinic through apheresis (similar to a red blood cell donation). Next, the patient materials are tagged clearly with patient information and either chilled or frozen and shipped from the clinic to a manufacturing facility. Upon arrival, the patient material is washed and select cells are isolated and then genetically modified. The genetic modification is typically performed using a viral vector (e.g., adeno-associated virus [AAV] or a lentivirus); however, other technologies—such as mRNA (or other RNA), cleavage enzymes (e.g., TALEN or CRISPR), and others (e.g., Cas-CLOVER, zinc finger)—are being explored. Once the genetic modification has been performed, the cells are expanded to a therapeutic quantity, concentrated, formulated, filled, and shipped back to the clinic for administration to the same patient.
This is obviously a complicated supply chain and requires close attention to track specific patient materials.7 Cold chain and traceability should be considered as part of the facility design, and special precautions should be taken to avoid the mixing of autologous materials. Many patients who are receiving these autologous treatments are incredibly sick, and they may only have time for one cycle that could last from three to four weeks. In this scenario, patient cells essentially become the patient. To drive that point home, some companies put the patient’s name on the cells throughout the process. When designing a cell therapy facility, it’s important to keep this larger picture in mind as well.
In the case of allogenic therapies, the process is similar, but the source materials vary. The major difference is that the starting cells come from donors or, in some cases, a cell bank. The therapy is not necessarily individualized to a single patient, and the same lot can potentially be used to treat a number of patients. The process resembles more of a straight line than a cycle, but cold chain and traceability remain important.
Case Study
For many emerging cell therapy companies, a popular strategy is to use a contract manufacturing organization (CMO) to handle the product through clinical trials in one facility and then transfer operations to a newly built manufacturing facility for commercial production. On a recent project, IPS worked with a CMO that was looking to expand and improve their cell therapy manufacturing spaces. contract manufacturing organizations need to be able to adapt to changing markets, customers, and customer needs. As such, the contract manufacturing organization project team had three major facility design goals:
- The facility will be flexible enough to handle multiple products.
- The facility can handle both autologous and allogenic products.
- The facility can handle both open and closed processing.
These goals are important aspects of designing a single facility for multiple cell therapy processes. Relevant questions include “What specific aspects of the unit operations need to be considered?” and “What process risks need to be considered?” To answer these questions, a strong understanding of what the processes might look like is needed. In this case, most processes likely fall within one of the following three categories:
- Straight cell expansion: A sample of cells, whether it be autologous or allogenic, is grown to a therapeutic dose. An example could be a stem cell therapy.
- Cell processing and filling facility: This operation is similar to a straight cell expansion, but there are some extra processing steps along the way. Considerations also need to be made for aseptic filling on site.
- Multistep aseptic microbiology process: This process involves intensive manual manipulation, with most steps performed within a biosafety cabinet (BSC).
Open versus Closed Processing
With regard to facility design, one of the biggest considerations of the manufacturing process is whether it operates in an open or closed fashion. Cells in an open process are exposed to operators and the surrounding environment, and high-grade air supply is required. Cells in a fully closed process are never exposed to the surrounding environment. On the other hand, functionally closed systems are routinely opened and then returned to a closed state via sanitization, cleaning, or sterilization prior to product contact. Closed processing is typically achieved with single-use pathways. However, isolators can be used where a single-use option does not exist.
Open Processing
According to US FDA guidance, 8 open aseptic processing should be performed in a Grade B environment (e.g., a BSC), which is typically in a Grade C surrounding environment. Concurrent open processing of different batches may occur in different workstations in the same suite by dedicated operators,9 but only under strict operational controls:
- The use of a suite must be based on campaigns (same viral vector).
- Workspaces must be operationally segregated.
- Operators must be dedicated to a workstation (no patient material crossflow).
- Only one patient batch can be used in each BSC.
- Proper line clearance/changeover procedures must be followed after each processing step.
These requirements cause big challenges in scaling the process out. The facility is limited by the number of BSCs and the segregation methods employed. While scale-up is an issue, open processing does come with a major advantage: there is little need for change between the laboratory-scale and commercial-scale processes.
From a facility design perspective, the following challenges need to be considered:
- The need to design processing suites for a Grade B background.
- The volume of material movement between BSCs, incubators, and other operating steps.
- Operator quantity and variability. With largely manual processes, the techniques of different operators vary enough that consistent product quality can be difficult to achieve.
- The difficulty of defining a boundary for cleaning and decontamination.
- Additional environmental monitoring (EM) requirements at the BSCs and for personnel and surfaces at the end of operations, batches, and/or campaigns.
Fully Closed Processing
Closed systems typically utilize single-use equipment and tubing sets; all material transfer is through presterilized tubes connected by sterile aseptic connectors, or with connections made by a tube welder. Safeguards such as HVAC design and room changeover procedures must be in place for operator safety and cross-contamination mitigation in case the system is breached. Scale-up to commercial scale is typically much easier in closed systems than in open ones, and closed systems are less reliant on operator technique. Although closed systems certainly involve a higher initial investment in terms of development time and equipment costs, they may reduce manufacturing costs because they have lower room classification requirements, require fewer operators, and have a smaller cleanroom footprint.
Functionally Closed Processing
Functionally closed systems operate in much the same way as fully closed systems, but they have one or more routine operations that require the process to be opened (installation of a filter, making a connection, etc.). Once the open operation is complete, the system is returned to a closed state by sterilization, sanitization, or cleaning before any product contact occurs. A risk assessment can be used to determine if and when open steps are at an acceptable risk level to be considered appropriate within a given environment.
Cell Therapy Technologies
The next part of the process that needs to be considered is what technologies are being used. This decision will drive whether the system will be open or closed. There are numerous suppliers for each unit operation, with some of the many vendors listed in Table 2.
One of the first considerations is whether an end-to-end cell processing platform will be used, or whether separate unit operations will be selected. End-to-end systems have advantages such as being closed and modular, but they can be relatively expensive, and, in some instances, having all unit operations combined can cause bottlenecks.
If separate unit operations are required, additional questions must be posed:
- Is an adherent or suspended cell culture being used?
- What is the scale of the cell culture process? This decision will drive whether a T-flask, cell stack, cell factory, or bioreactor makes sense.
- What transfection technologies are being employed? Certain technologies, such as flow electroporation, are currently incompatible with closed pro-cessing.
- Is a stabilizer added before product filling and freezing? If so, will it necessitate a quick formulation, filling, and freezing operation?
- Is a filling step required as part of the process? Will an open vial or closed vial technology be used? How many containers need to be filled? Will it be a robotic filling operation or a manual operation?
These decisions will drive containment and facility segregation requirements. Understanding the process equipment intricacies and limitations is essential to ensure the facility is designed with enough flexibility to handle each scenario. Bear in mind that this list of questions is not all-encompassing, and each process must be examined thoroughly to determine specific requirements.
Managing Risks and Biosafety
Many traditional biologic therapies, such as monoclonal antibodies or recombinant proteins, are small enough that they can be sterile filtered (0.2 μm) prior to filling. That is not the case with cell therapies, as they cannot be terminally sterilized. Because of the relatively large size and fragility of cell therapies, typical sterilization techniques such as filtration, heat, or ultraviolet light are not options. It is easy to forget these therapies are living cells. Therefore, aseptic processing is required to ensure product safety, especially for injectable drugs, and the use of closed manufacturing systems is encouraged wherever possible.8
| Area of Operations | Technology/Operating Principle | Vendor/Technology Examples |
|---|---|---|
| Leukapheresis/apheresis | Centrifugation | Terumo BCT: Spectra Optia Blood Bank Equipment |
| Cell processing | Immunomagnetic | Miltenyi Biotec: CliniMACS Plus Miltenyi Biotec: CliniMACS Prodigy AdvaBio: Adva X3 |
| Microfluidic | GPB Scientific: Curate | |
| Centrifugation | Cytiva: Sefi a S-2000 Cytiva: Sepax 2 Cytiva: Sepax C-Pro Fresenius Kabi: Lovo Terumo BCT: ELUTRA Terumo BCT: COBE |
|
| Electroporation | MaxCyte: GT, STX, or VLX Lonza: Nucleofector |
|
| Acoustic cell retention | Draper (system name to be determined) FloDesign Sonic (Millipore Sigma): ekko |
|
| Immunodensity cell separation | Reagent based with centrifugation | |
| Adherent cell culture | 2D | Corning: Cellcube Corning: HYPERStack ThermoFisher: Nunc Cell Factory Wilson Wolf: G-REX Terumo BCT: QUANTUM Pall: Xpansion Cytiva: Xuri |
| 3D matrix | Octane Biotech: Cocoon Pall: iCELLis |
|
| Microcarriers | Pall: SoloHill Cytiva: Cytodex |
|
| Freezing/thawing | Controlled rate freezing/thawing | Cytiva: VIA ThermoFisher: CryoMed Sexton Biotechnologies: Cell Seal Thaw |
| Filling | Open | Sexton Biotechnologies: Cell Seal AF-500 Sexton Biotechnologies: Cell Seal SAFS-100 Aseptic Technologies: M1 Aseptic Technologies: L1/SL1 |
| Closed | Terumo BCT: FINIA Sexton Biotechnologies: Signata CT-5 |
|
| Cell storage | Liquid nitrogen | Cryotherm Brooks Life Sciences |
Cold and cryogenic shipment, receiving, and tracking are very important to maintain starting material viability. The expression and nature of biomolecules and factors that are present in apheresis collections for cell therapies are not currently well defined and measurable; as a result, these materials are fragile and sensitive to changes in cold chain procedure. Other dangers include humidity, shock, and carbon dioxide ingress, which, in addition to temperature, could all have an effect on cell metabolic function during transportation. Traceability is also essential to ensure that products are supplied and administered to the correct patients. Facility automation and integration can help alleviate some of these challenges and reduce supply chain risks.
| BL | Practices | Primary Barrier and Equipment | Facilities (Secondary Barriers) |
|---|---|---|---|
| 1 | Standard microbiological practices | None required | Laboratory bench and sink required |
| 2 | BL1 practices plus:
|
Primary barriers:
|
BL1 facilities plus:
|
| 2-Plus | BL2 practices plus one or more of:
|
Primary barriers:
|
BL2 facilities plus:
|
Biosafety of the facility needs to be evaluated during a biological risk assessment.10 The risk assessment evaluates agent risk to both personnel and the environment, procedure risk, and appropriate risk management measures. Risk groups are an assessment of the agent, and biosafety levels (BLs) are the appropriate methods to control the risk (see Table 3). The NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules11state, “The institution shall appoint a Biological Safety Officer if it engages in large-scale research or production activities involving viable organisms containing recombinant or synthetic nucleic acid molecules.” In this instance, large scale is defined as volume greater than 10 liters.
The agents that need to be considered with regard to biosafety are donor materials, cells from cell banks, and viral vectors. Donor materials need to be prescreened for bloodborne pathogens, or the facility must be designed to handle biohazardous materials. Cell banks need to be evaluated for biohazard level because some cell lines may contain carcinogenic materials. Additionally, if any viral vectors will be used during cell processing, the vectors must be assessed for biohazard level. It is common for cell therapy facilities to be designed to a BL2-plus level. However, BL2-plus is not actually a prescriptive safety level in the NIH guidelines;11 rather, BL2-plus meets more than the elements of BL2, but less than those of BL3. These improvements are usually with regard to facility directional airflow or additional containment, which are not requirements to meet BL2.
Cross Contamination
The single greatest driving factor for cell therapy facility design is the mitigation of the risk of cross-contamination. What constitutes cross-contamination? During cell therapy processes, the goal is to grow a single cell line. Contamination of another cell or viral vector from a different process or source could cause major problems in culture mediums, and these contaminations could seriously compromise the quality of the therapeutic product, potentially causing tumorigenic and immunogenic risks in patients if the contamination is not discovered. This is different than the other contamination risks considered in aseptic operations, such as bacterial and fungal contaminations.
As touched on previously, some of the major ways to prevent cross contamination are closed systems and single-use technologies. Other preventive measures involve risk mitigation associated with personnel, materials, waste, and product flows. The gold standard is unidirectional flows in a facility, where raw materials start at one end and products and waste come out the other. This is not always achievable because of the orientation of an existing space or economic constraints. Expenses such as a separate entrance and exit airlocks for processing suites, separate supply and return corridors, and so on can add up quickly. If perfect unidirectional flow is not achievable, a risk assessment is required to determine where the risk of deviating from unidirectional flow is small enough to be acceptable.
Another essential aspect of cross-contamination that needs to be considered is HVAC design. Although the number of air changes present in Grade B cleanrooms (ISO 5 in operation) is higher than in Grade C cleanrooms (ISO 7 in operation), that does not necessarily mean that cross-contamination risk is lower in a Grade B environment. There are two major concepts to address: (a) isolation, where the focus is on keeping all the process components and materials within one architectural suite, and (b) segregation, where the focus is on separating facility air-handling units (AHUs) depending on risk assessment.
Isolation
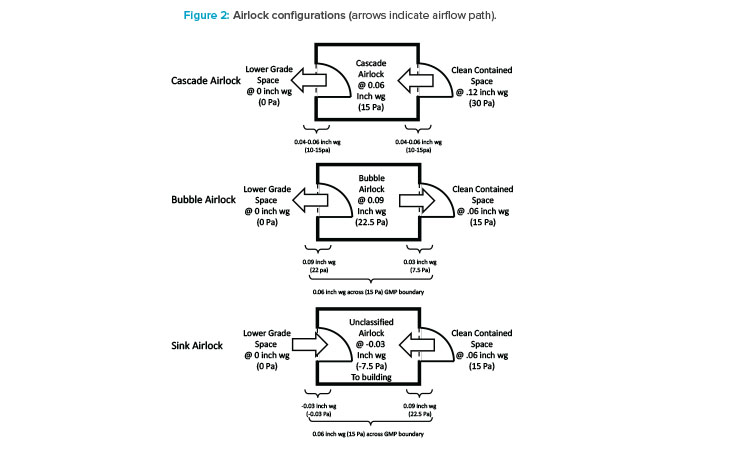
First, considering isolation, potential contaminants are likely coming from outside the processing suite, so the room boundaries, which are airlocks, need to be considered (see Figure 2). Traditionally, the goal of airlocks is to keep cleanrooms clean, so clean air cascades across an airlock flowing from the clean side to the dirty side. In a multiproduct facility, a cascade design could potentially lead to the contamination of a shared hallway with the cellular or viral particles of a process. For this reason, a bubble and sink design is the best airlock design for multiproduct facilities. Bubble airlocks are pressurized to provide clean air to both sides of the airlock at the entrance of a cleanroom. Sink airlocks, on the other hand, pull air from both the cleanroom and shared hallway at the exit. The goal of this configuration is to isolate processing suites from one another. This is necessary for two reasons:
- To contain products, media, and components of a product, preventing them from contaminating any other suite.
- To prevent any bioburden outbreak that might occur in one suite from traveling into another suite, whether it is used for a similar product or not. Isolation facilitates quick identification and remediation and prevents plant-wide contamination.
Segregation
When considering process segregation, the risk assessment centers on how the failure of any single AHU affects the rest of the facility. Designs should ensure that failure of one AHU only affects a single product/processing suite. This means multiproduct facilities should not share supply and return air ducts between the suites so as to prevent cross-contamination between suites in the event of a spill or bioburden excursion. Cleanroom designs are thus pushed to use smaller, less-expensive variable frequency drive-based AHUs, rather than the larger, more traditional style.12 An alternative is the consideration of once-through air supply. Although once-through air designs are costly in terms of energy efficiency, a return on investment calculation should be performed to determine if the reduced capital expenditure and increased environmental impact for a once-through design outweighs the energy savings of a recirculated design.
Room changeover and cleaning methods need to be considered when changing from one product campaign to the next. One method that is being used more frequently is vaporized hydrogen peroxide (VHP). Although other sterilizing vapors such as chlorine dioxide have been considered, VHP is the most commonly applied because of its lower relative humidity required. This method is frequently used in isolators to kill potential contaminants, and it can also be used to change over rooms using fixed or mobile equipment. VHP tends to stick around, absorbing into plastics, only to be off-gassed slowly over time. This could potentially lead to lower product yields for cell therapy processes if the VHP is not sufficiently removed from the room after the decontamination cycle. It is for this reason that careful attention must be paid to HVAC design and target residual VHP levels.
Facility Automation
There are three major challenges that cell therapy companies face with regard to manufacturing and delivering their products to patients:
- Manufacturing productivity
- Achieving compliance
- Scalability
Specially designed facility automation platforms can help address these issues. Manufacturing productivity can be improved by integrating clinical treatment facilities, delivery logistics, manufacturing execution systems, enterprise resource planning, and customer resource management systems. Rather than relying on disjointed manual processes, integration allows for a trackable, digital workflow allowing for easier scheduling.13 Additionally, materials and products could be tracked by barcodes or wireless radiofrequency identification technology throughout the process. Facility automation can also limit particulate generation in aseptic processing, where an operator’s manual manipulations would normally take place. In the same vein, integration allows for stronger compliance as well.
It is possible that a record of continuity throughout the process may become a requirement for compliance. While compliance reviews for tradition-al therapies can last a month or more, processing for autologous therapies must be completed in days. The need for expedited batch record review and approval is obvious.
In addition to the aspect of improved compliance, reliability of the logistics supporting the cold chain and traceability are strengthened, minimizing the risk of mix-ups (e.g., delivering the wrong cells to a patient). Scalability is the biggest issue facing many cell therapy processes, and logistics is the biggest hurdle with regard to scalability. Automation integration can help with this aspect of the facility as well.14 Having a cloud-based platform that provides accessibility of data not only to the manufacturing facility operations team but also to cell therapy customers and partners allows for better communication and can reduce logistics development and deployment time.
Layout Considerations
There are a multitude of options and opinions on how cell therapy facilities are organized and laid out. Flexibility and modularity are universal concepts in designs, but how these aspects are approached can differ. Modules are typically designed as “ballrooms” where a series of suites may lead to one large multipurpose room, or all of the processing may take place in the single ballroom. Many cell therapies have separate upstream and downstream modules. While the ballroom design is more typical of research and development and clinical production, a modular approach allows for scale-out. As incremental production demands change over time, from clinical trials to commercial production, additional processing modules are added. Traditional design and building methods are able to meet the basic requirements of the cell therapy facilities, but other methods, such as modular wall construction or prefabricated modular delivery, are often better choices to meet highly compressed project timelines.
Flexibility is key when designing modular layouts, especially for a CMO that will be hosting different customers with different needs. Figure 3 shows different configurations for multiple cell therapy suites that were designed for Grade B operations and flexible enough to handle multiple process con-figurations. Specifically, process gases and chilled water were provided to support many different equipment configurations. Process gas utility panels are typically supplied on the ceiling for maximum flexibility, while the chilled water is supplied at the walls.
Special considerations should be made for equipment selection with regard to structural elements. For example, some incubator designs require a penetration through an architectural wall, so maintenance access is available from a mechanical space adjacent to the cleanroom. This type of incubator design is not amenable to a flexible suite layout because of the architectural changes required. Suites 1 and 3 are configured for a two-dimensional (adherent) cell culture operation that uses open operations in T-flasks and cells stacks and heavily relies on operator manipulations in BSCs and incubators. Suite 2 is configured for a three-dimensional (suspended) cell culture operation that utilizes closed single-use bioreactors. For a closed system, a Grade B background is not required; therefore, designing flexibility into the HVAC system so that it can be requalified to a lower grade should be a consideration.
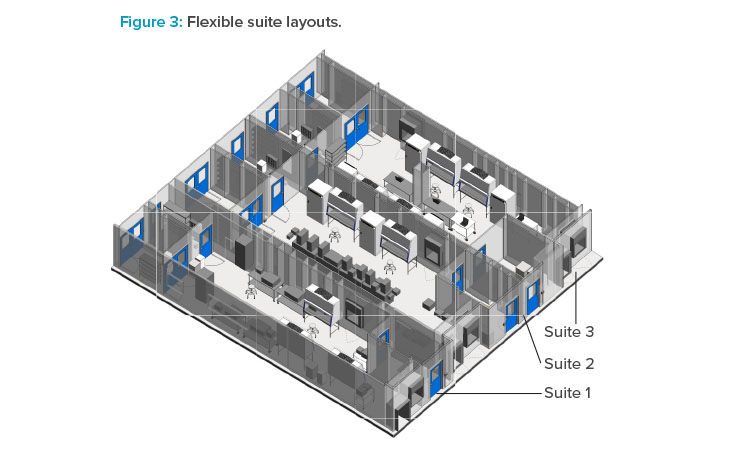
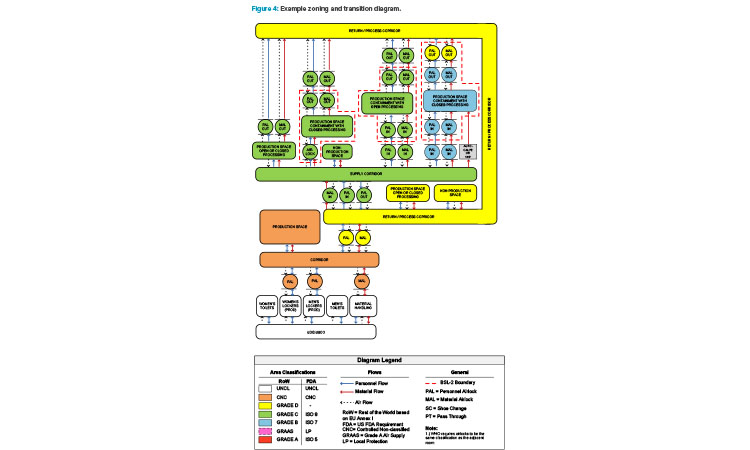
Figure 4 is an example of a zoning and transition diagram showing the personnel flows, material flows, and airflows for several processing suite con-figurations within the context of a facility. Looking at the diagram from left to right, both flexibility and cost to build increase. A strong understanding of the underlying requirements of the facility and potential processes is necessary to choose a layout that has the right amount of flexibility while minimizing unnecessary expense. Note the “bubble” airlock entrances and “sink” airlock exits to the production suites and how these line up with potential biosafety boundaries (dashed red lines).
Case Study Review
Now that layouts that fit within the context of the rest of our facility have been established, it’s time to review the goals established in our CMO case study and how they have been addressed.
- The facility is flexible enough to handle multiple products:
- Biosafety has been considered in the HVAC design, waste flows, and decontamination steps.
- Cross contamination has been addressed with the use of closed process technologies, single-use equipment, HVAC design, and room changeover procedures.
- Utility panels are provided in optimized locations to support future flexibility.
- The facility can handle both autologous and allogeneic processes:
- The automation designs considered make robust traceability and cold chain possible for autologous products.
- Materials flows have been considered and are optimized for unidirectional flow as much as possible.
- Support rooms are designed in such a way that they allow for optimized material and personnel flows.
- The facility can handle both open and closed processing:
- A Grade B suite is capable of working with both open and closed processes and can be designed to be requalified to Grade C, if necessary, in the future. If optimum flexibility is required, as in the case of our CMO, the suites must be designed to Grade B. If less flexibility is acceptable, a mix of Grade B and Grade C suites can be considered.
Although not every cell therapy process is amendable to this type of flexible facility design, the principles are still applicable in most situations, and especially in those cases where multiple products are manufactured in the same facility.
About the Author



