Extractables & Leachables: Not the Same

With the integration of single-use systems (SUS)* into downstream processing and thus closer to the final drug product, considerations of extractables and leachables (E&L) have become a critical issue within the industry. Lack of standardization, however, leads to incomplete E&L studies that do not cover the conditions encountered throughout the process train. This makes it difficult for end users to select suitable single-use components.
The pharmaceutical industry’s challenges are:
- Prevent misinterpretation of regulatory requirements for E&L as they are used on finished product containers by applying them to process contact materials as well
- Bridge the gaps between end user expectations and supplier capabilities by defining the limit of responsibilities and the scope of operations for both.
The objective of this article is to clarify and highlight the importance of distinguishing between extractables and leachables when evaluating SUS. Our goal is to propose consensus E&L guidelines for all industry stakeholders, using risk management and quality by design (QBD) approaches, and supporting single-use development and market promotion.
This paper presents risk-based approaches for evaluating E&L from SUS. In fact, regulations on single-use-technologies (SUT) do not exist yet, although the basis for such regulation exists. In the United States, for instance, 21 CFR 211.65 states, “Equipment shall be constructed so that surfaces that contact components, in-process materials, or drug products shall not be reactive, additive, or absorptive so as to alter the safety, identity, strength, quality, or purity of the drug product beyond the official or other established requirements.” 1
TWO WORDS, TWO PERSPECTIVES
SUT has been part of the biopharmaceutical industry for about a decade. Because terminology is a key element in preparing and understanding any new area, it is of crucial importance to define common terms and use them properly.
Many people use “extractables and leachables” as a single term, but these concepts reflect two very different chemical species, although both migrate from the component.
Extractables are chemical compounds that migrate from SUS into model solvent solutions under controlled and exaggerated conditions depending on temperature, pH, polarity, and time. SUS are normally not exposed to such conditions in biopharmaceutical processes.
Leachables are chemical compounds that migrate from SUS into process solutions under normal biopharmaceutical process conditions; they may end up in the final drug product formulation. For the most part, leachables are a subset of extractables, although interaction with product components may produce leachables not seen as extractables.
Extractable and leachable studies pursue different objectives: Extractable studies are designed to obtain a fingerprint of chemical components that can be extracted under exaggerated conditions. Toxicological review of these fingerprints and risk assessments for potentially problematic components helps select appropriate SUS. Extractable studies can also be used as a baseline to ensure SUS consistency over time.
Leachable studies determine the chemical compounds that migrate from SUS into process solutions and characterize possible adsorption and/or absorption of process fluid (under normal process conditions). This data enables a toxicologist to determine if components that can compromise patient safety are present in the drug product. In addition, leachables data can indicate the presence of chemical components that could potentially interact with the drug product itself, and can help assess the potential for alterations of the drug potency and/or stability. Since leaching continues over time, posing a risk to patient safety and drug product efficacy, leachables may also appear in stability studies.
These definitions (especially for extractables) are not totally balanced. Different applications and situations in biomanufacturing process must be categorized to highlight the weight and criticality of both E&L profiles in product contact material.
COLLABORATION
Regulatory authorities expect that the final dosage form will have a well-characterized degradant profile, including leachables from process contact materials, even if they do not pose any major health risk for the patient. As part of this profile, E&Ls must be targeted, assessed, and mitigated. This is challenging due to the number of parameters that can affect E&L evaluation and the need for trace-level analysis.
Collaboration and clear definition of roles between SUS manufacturers, suppliers, and end users (drug product manufacturers) are crucial to ensuring patient safety and product efficacy. This is true even for the transportation and storage of single-use components and assemblies prior to use in the drug product manufacturing process.
The easiest way to determine responsibilities is to establish clear communication and transparent exchange of documentation (certification, report, conclusion, minutes of decision meeting, and any legal agreements). Tools such as a RACI (responsible, accountable, consulted, informed) responsibility assignment table could be used to determine stakeholder responsibilities.
* Presterilized products, equipment, and packaging designed to be used once or a few times, depending on specific circumstances, and discarded.
RISK-BASED APPROACH
A risk assessment for both extractables and leachables that balances business risk and patient safety should be established for different phases of SUS manufacturing and drug development. Business risk must be considered, but never at the expense of patient safety. Suppliers have considerable commitment to the cost of extractable analysis with regard to end user expectations (user requirement specifications) and the feasibility of conducting a more complex extractables study.
Both end users and suppliers have a stake in the business impact analysis. Ultimately, the main consideration must be on product quality and safety, which should be assessed with appropriate risk assessment and mitigation strategies (such as quality risk management). The amount of leachables per unit of final drug product dosage form (along with posology) is the final regulatory expectation. End users must comply with this patient-risk approach as required by regulatory bodies.
QBD APPROACH
An E&L program should be based on QBD principles and a thorough understanding of the biomanufacturing process. Using this approach, suppliers should conduct E&L studies on in-process SUS from sourcing of raw materials to disposal—including key milestones such as sterilization—in compliance with baseline safety assessment and chemical studies. End users should first evaluate the criticality of the SUS component regarding process flow, based on documentation provided by the supplier.
Chemical fingerprint analyses will ensure that no toxic substances are found (or are well below the limit) and that the product is unlikely to interact with the final drug product. Controlled extraction studies are needed to make an informed selection of materials, meet regulatory expectations, evaluate safety of materials, and control leachables absorbed in the final dosage form.
Many people use “Extractables and Leachables” as a single term, but these concepts reflect two very different chemical species, although both migrate from the component
EXTRACTABLE EVALUATION METHODOLOGY
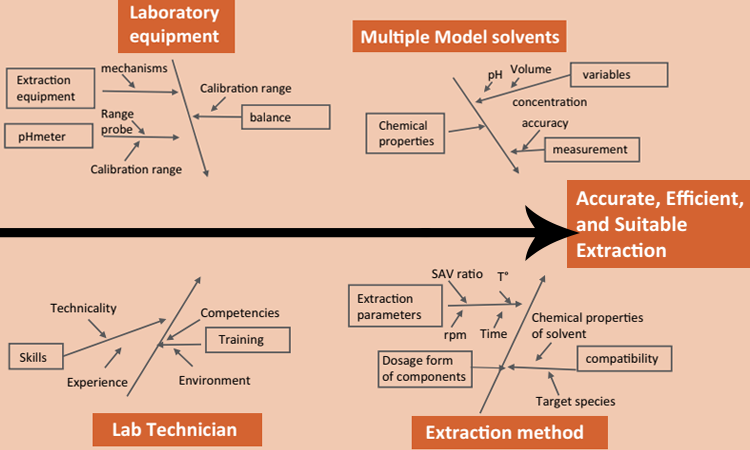
Before conducting an extractables study on an in-process SUS, end users should assess single-use material attributes—dosage form, formulation composition, intended use, and stability—within a risk-assessment approach, considering the intended use and process conditions of the SUS component. The experiment design should consider several factors (Figure 1) that could affect the quality of extraction and resulting analysis:
- Model solvents selection
- Surface area to volume ratio
- Mass of each extractable or leachable per volume of model solvent
- Time points of extraction
- Type of material tested
- Related structural and physical properties: SUT resin, film, component, assembly, and system
Components should be tested using multiple extraction techniques and solvents of various polarities according to a wide range of targeted species and dosage forms. Because “most” does not mean “better,” one should not expect a maximal number of chemical compounds during contact material extraction (time of contact, concentration of solvent, extraction kinetic) butrather select appropriate extraction conditions and analytical approaches predictive for leachables produced in a specific application. The extract is then evaluated with analytical techniques based on sensitivity, limit of detection, and the target species properties (considering first volatiles, semivolatiles, or nonvolatiles).
Following a toxicological analysis, the extractable can be identified and evaluated for its potential toxicity and safety threshold. If potential toxicity is discovered, end users should report the results to suppliers and stop using the SUS component tested until a risk mitigation strategy has been enacted. Suppliers should follow common extraction methodology based on standards recognized by all industries, and communication to end users must be based on consensual reporting methods.
- 1US Food and Drug Administration. Code of Federal Regulations, Title 21, Chapter I, Subchapter C, Part 211, Subpart D, Sec. 211.65, “Equipment Construction.” https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=211.65
LEACHABLE EVALUATION METHODOLOGY
Evaluation of SUS leachables should be based on a risk- and science-based approach to ensure the safety and purity of the final drug product. Process knowledge, experience gained during development, and a comprehensive process understanding should be used to assess risk associated with implementing SUS. Risk management principles can identify, evaluate, communicate, and mitigate leachables that can affect product quality and patient safety. A leachable profile should be used to determine the residual chemical identity of the SUS in normal process conditions and the toxicological impact on drug product and on patient safety. A leachable material is objectionable if it adversely affects critical quality attributes such as purity, safety, efficacy, identity, strength of the final and/or intermediate product, or its successful production.
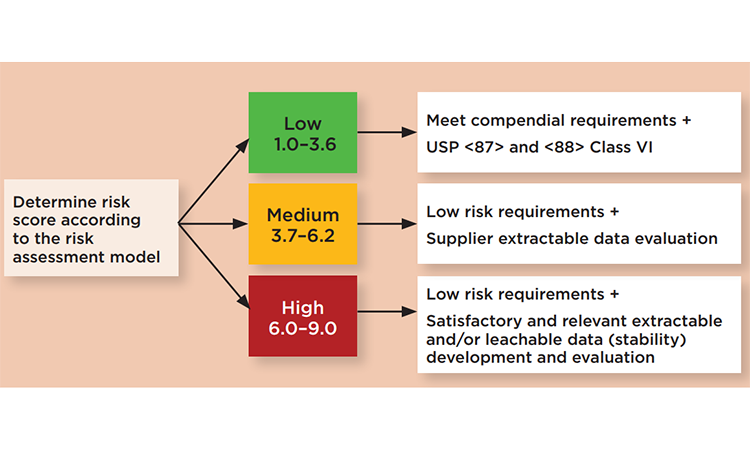
Risk may be based on severity of the harm caused by leachates from SUS, probability that leaching will occur, and probability of detecting the leached substances through in-process manufacturing controls. Once the overall risk rating of the single-use component of interest is finalized and ranked (low, medium, or high), qualification requirements should be established to qualify the single-use component for its intended use (Figure 2).
We recommend a case-by-case approach to define which extractables should be analyzed in a leachables study. For medium and higher rated risks, product-specific assessments should be based on extractable data and conducted under a toxicologist’s supervision. Given high-quality data that is applicable to the end user bioprocess, extractables data can guide and define the depth of a leachables study. In addition, end users should consider that leachables may also be derived from the interaction between an extractable and a drug formulation compound.
CONCLUSION
A key advantage of implementing this approach for process contact materials is that it identifies where a leachable study is needed, and focuses the effort according to leaching propensity. QbD risk assessment and synergetic collaboration between suppliers and end users are leading principles that should facilitate SUS implementation by providing better understanding of industry expectations for suppliers. This drives uniformity of study design, allowing integration of data from multiple suppliers, and facilitating evaluations and comparisons between components.
Acknowledgments
Malik Belattar thanks coauthors Nassrine Lablack, Quality and Regulatory Compliance Engineer at the SUTAP organization, and Mathieu Tricot, Quality and Regulatory Engineer at Pharmabiot’Expert, for their technical assistance and contribution for drafting the article. He is also grateful to Christopher J. Smalley, Director at Merck & Co., and Sabrina Restrepo, Associate Director, Component Engineering at Merck, for their valuable insights and technical review of this paper.


