Managing Potential Virus and TSE Contamination

Biopharmaceutical products manufactured from mammalian or microbial cells are inherently at risk of contamination from two major types of entities.
A Risk-Based Approach for cGMP Biopharm Manufacturing Facilities
The first type consists of adventitious or endogenous viruses. Viruses are composed of a nucleic acid genome surrounded by a proteinaceous capsid, with or without a lipid envelope requiring a host cell for replication. Adventitious viruses are introduced to the manufacturing process unintentionally; endogenous viruses are already present in the cell line and may be part of the host genome.
The second type consists of transmissible spongiform encephalopathy agents (TSE, or prion diseases), which can be introduced into the manufacturing processes by contaminated ruminant (cattle or sheep) materials. TSEs are neurodegenerative disease-causing agents that contain no genetic material. TSEs affect humans and animals, and are characterized by the accumulation of an abnormal isoform of a cellular glycoprotein known as PrP, or prion protein. Prions are highly resistant to physicochemical inactivation procedures such as heat, ionization, ultraviolet light, microwaves, irradiation, and acid treatment. Reducing TSE infectivity risk relies on stringent methods such as treatment with a strong base and the elimination of animal-derived raw materials (ADRMs) from the manufacturing process. Both viruses and TSEs pose a risk to the entire manufacturing facility.
Viruses
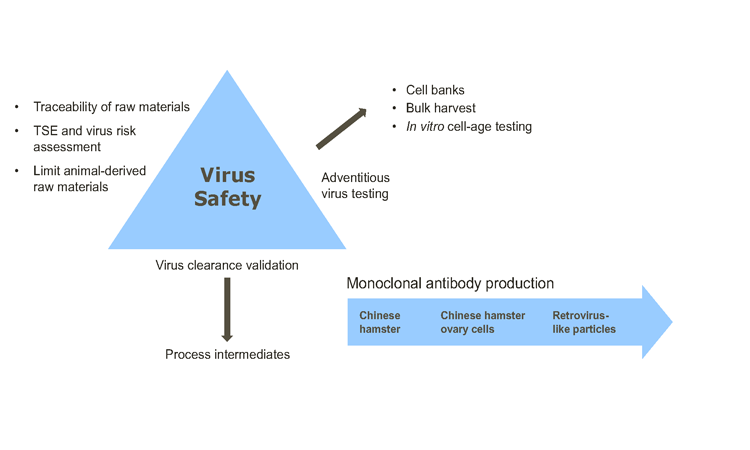
Virus safety assurance for biopharmaceuticals derived from cell lines of human, microbial, or animal origin is demonstrated by a threefold approach described in ICH,1 FDA,2 ,3 and EMEA4 regulatory guidelines (Figure 1).∗ The key virus safety components are:
- Selecting, assessing, tracing, and testing cell lines and raw materials (including media components) for the absence of viruses and limiting the use of ADRMs;
- Testing the product for the absence of viruses at appropriate stages of production; and
- For mammalian cells, assessing the capability of production processes to clear infectious viruses.
TSEs
Bovine spongiform encephalopathy (and its link to variant Creutzfeldt-Jakob Disease,6 which was first identified in the UK in 1996) has had a significant impact on the biopharmaceutical industry. As a result, international agencies—including those in Europe, the United States, and Japan—have published guidelines applicable to the management of TSE risk to minimize patient exposure via pharmaceutical products.5 ,7 ,8
While the risk of TSE propagation by mammalian cells is low,9 measures to eliminate ADRMs must be enacted to mitigate potential exposure. If this is not possible, a comprehensive risk assessment based on species, tissue, country of origin, and manufacturing process used to produce the raw material or component5 should demonstrate that there is no residual risk from TSE agents.
Risk-management Program
These and other quality and regulatory expectations can be managed through a comprehensive virus and TSE risk-management program and control strategy. Examples include the implementation of a current good manufacturing practice (cGMP) philosophy and personnel training that encompasses all the operations carried out in the manufacturing facility.
Other considerations include:
- Facility controls such as ISO room classifications; heating, ventilation, and air-conditioning (HVAC) systems; laminar flow hood use; closed processing; process segregation; cleaning procedures; and pest control
- Equipment use and maintenance
- Equipment type, i.e., single use vs. stainless steel
- Personnel, waste and material flow strategies
This holistic approach to managing virus and TSE risk provides confidence that the clinical materials supplied to patients are free from the risk of contamination.
“Both Viruses And TSEs Pose A Risk To The Entire Manufacturing Facility”
∗ ICH: International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; FDA: US Food and Drug Administration; EMEA: European Medicines Agency
- 1International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline. “Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin: Q5A(R1).” 23 September 1999. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5A_R1/Step4/Q5A_R1__Guideline.pdf
- 2U.S. Department of Health and Human Services. “Points to Consider in the Characterization of Cell Lines Used to Produce Biologicals.” 12 July 1993. Memorandum docket 84N-0154.https://www.fda.gov/downloads/BiologicsBloodVaccines/SafetyAvailability/UCM162863.pdf
- 3U.S. Food and Drug Administration. Center for Biologics Evaluation and Research. “Points to Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use.” Docket No. 94D-0259. 28 February 1997. https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/OtherRecommendationsforManufacturers/UCM153182.pdf
- 4European Medicines Agency. “Guideline on Virus Safety Evaluation of Biotechnological Investigational Medicinal Products.” EMEA/CHMP/BWP/398498/2005. 24 July 2008. http://www.ema.europa.eu/docs/en_GB/document_library/Scientifi c_guideline/2009/09/WC500003795.pdf
- 6Worth Matravers, Lord Phillips of (Nicholas Phillips), June Bridgeman, and Malcom Ferguson- Smith. The BSE Inquiry Report Vol. 1: Findings and Conclusions. UK National Archives, October 2000. http://webarchive.nationalarchives.gov.uk/20090505195026/http://www.bseinquiry.gov.uk/report/index.htm
- 5 a b European Commission. “Note for Guidance on Minimizing the Risk of Transmitting Animal Spongiform Encephalopathy Agents via Human and Veterinary Medicinal Products (EMA/410/01rev.3).” Ocial Journal of the European Union, C 73 (2011). http://www.ema.europa.eu/docs/ en_GB/document_library/Scientific_guideline/2009/09/WC500003700.pdf
- 7US Food and Drug Administration. Center for Biologics Evaluation and Research. Letter to Manufacturers of Biological Products. 19 April 2000. https://www.fda.gov/downloads/BiologicsBloodVaccines/SafetyAvailability/UCM162938.pdf
- 8Ministry of Health, Labour, and Welfare. “Partial Revision of the Standards for Biological Ingredients.” Notification no. 375. 26 September 2014.
- 9Piccardo, P., et al. “Candidate Cell Substrates, Vaccine Production, and Transmissible Spongiform Encephalopathies.” Emerging Infectious Diseases 17, no. 12 (December 2011). https://wwwnc. cdc.gov/eid/article/17/12/11-0607_article
Control Framework
The risk of virus contamination in manufacturing facilities is also low, but when it occurs, the impact is severe. Examples of facility contamination recorded in the literature10 , 11 ,12 describe economic losses sustained in facility recovery after an incident, and patient hardship caused by loss or interruption of drug supply.
Viruses can infect mammalian production cells such as Chinese hamster ovary cells13 and NS0 cells (derived from a non-secreting murine myeloma). Biopharmaceuticals derived from Escherichia coli are also at risk from endogenous14 and adventitious bacteriophages.15 Viruses of yeast—both retroviral-like elements such as Ty1† 16 and double-stranded RNA viruses—may be transmitted during mating, but do not appear to produce particles that are infectious via an extracellular route.
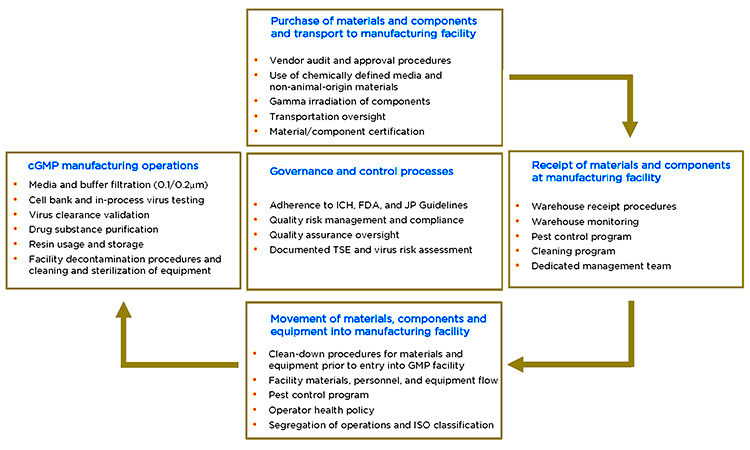
The control of risk is governed by ICH Q9 guidelines17 applied to areas with the potential for TSE and virus contamination (Figure 2). These areas include raw material and component sourcing, transport, and receipt; movement of materials into the manufacturing facility; buffer and media makeup; and GMP manufacturing operations. Requirements for virus and TSE control are also outlined by regulatory authorities and requirements in internal policies, procedures, and controls—including facility audits.
In virus and TSE risk assessments, each manufacturing step is reviewed for the risk of contamination from personnel, material, and environmental sources. Risk values of probability and occurrence are assigned both before and after mitigation actions are taken. Segregation strategies and general risk-mitigation steps are also reviewed.
Raw Materials And Components
The main sources of risk for virus contamination are ADRMs or animal-derived ingredients (ADIs). TSE contamination may arise from tissues or secretions of animals—primarily ruminants—susceptible to prion diseases used to prepare cell banks or medicinal product. Primary ADIs or ADRMs may be included as active substances, excipients, or adjuvants (fetal bovine serum, sheep-wool-derived cholesterol, or milk-derived galactose). Secondary or tertiary ADIs include recombinant proteins (e.g., insulin) manufactured in media that contain a ruminant material (such as milk) and/or are used as biopharmaceutical production media additives.
Ruminant substances (e.g., tallow) in product-contact materials (components, consumables, and equipment) must be controlled by identifying the species and tissue source, country of origin, and method of treatment during manufacture (alkaline hydrolysis and/or heat).
All materials must have supporting vendor documentation, such as a Certificate of Origin or European Directorate for the Quality of Medicines Certificate of Suitability.‡ This applies to materials used in cell lines prior to the manufacture of cell banks, GMP cell banks, and raw and product-contact materials used throughout the product life cycle. Measures should also be in place to prevent exposing process-contact equipment and utilities to materials of human or animal origin.
† Long-terminal repeat retrotransposon of the yeast species Saccharomyces cerevisiae, whose structure is similar to retroviruses
‡ A process for periodic renewal of these certificates via vendor management programs should also be in place.
- 10Garnick R.L. “Raw Materials as a Source of Contamination in Large-Scale Cell Culture.” Developments in Biological Standardization 93 (1998): 21–29.
- 11Sanofi Genzyme. “Genzyme Reports Progress Related to Allston Plant.” Press release. 25 June 2009. http://news.genzyme.com/press-release/genzyme-reports-progress-related-allston-plant
- 12Chen, J., et. al. “Case Study: A Novel Bacterial Contamination in Cell Culture Production—Leptospira Licerasiae.” PDA Journal of Pharmaceutical Science and Technology 66 (2012), 580–591.
- 13Wiebe, M.E., et al. “A Multifaceted Approach to Assure that Recombinant tPA Is Free of Adventitious Virus.” In Advances in Animal Cell Biology and Technology for Bioprocesses, edited by R. E. Spire. London: Butterworths, 1989.
- 14Shuman S. “Vaccinia DNA Topoisomerase I Promotes Illegitimate Recombination in Escherichia Coli.” Proceedings of the National Academy of Sciences 86 (1989): 3489–3493. http://www.pnas.org/content/86/10/3489.full.pdf
- 15Los, M., et al. “Bacteriophage Contamination: Is There a Simple Method to Reduce Its Deleterious Effects in Laboratory Cultures and Biotechnological Factories?” Journal of Applied Genetics 45, no.1 (2004): 111–120. http://jag.igr.poznan.pl/2004-Volume-45/1/pdf/2004_Volume_45_1-111-120.pdf
- 16Feng, Y-X, et al. “The Genomic RNA in Ty1 Virus-Like Particles is Dimeric.” Journal of Virology 74, no. 22 (2000): 10819–10821. ,a href="http://jvi.asm.org/content/74/22/10819.long">http://jvi.asm.org/content/74/22/10819.long
- 17International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline. “Quality Risk Management:Q9.” 9 November 2005. http://www.ich.org/fi leadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step4/Q9_Guideline.pdf
Transportation from the vendor to the site of manufacture provides another element of risk that must be considered. Moving materials from the warehouse into the GMP core should be governed by strict procedures that include disinfectant; materials, personnel, and equipment flow segregation; ISO room classification; and operator health policy. Cell banks should only be released for manufacture after a TSE and virus risk assessment, appropriate virus testing and approval of a written report.
Each of these steps helps ensure that the origins of all animal-derived raw and starting materials used to develop cell lines and manufacture biopharmaceutical products have been investigated, and that potential contamination risks from TSE or viral agents have been fully defined and assessed.
Manufacturing Facility Control
Manufacturing facility control incorporates GMP policies and procedures relevant to the facility operations as well as regulatory guidelines. HVAC systems aid in virus control by delivering terminal high-efficiency particulate (HEPA) filtered airflow to maintain segregation between zones, creating directional flow and differential pressures to provide the requisite number of air changes for classified rooms (ISO-5, ISO-7, ISO-8,). HVAC systems also prevent particulate material from migrating between processing areas and circulating or accumulating within rooms. Microbiological and particulate monitoring verifies the integrity of controlled environments and area classification requirements.
During purification operations, segregation through architectural design or by closed systems are important in pre and post-virus-removal steps. A system of airlocks prevents cross-contamination during entry to and exit from rooms and areas. Pass-throughs are kept clean and free of debris. All cell lines are tested for virus and risk-assessed for TSE, then dispositioned prior to use as required by standard operating procedures. Procedural controls are in place in the event of a facility contamination or pest ingress. Mitigating actions can be triggered by a positive (or false positive) adventitious and/or endogenous virus test.
“The Main Sources Of Risk For Virus Contamination Are ADRMs Or ADIs”
Equipment Considerations
As the industry moves toward closed processing, single-use bioreactors and chemically defined media (which must be free of ADIs) are also risk-reduction steps in TSE and virus control.
Single-use bioreactors (SUBs) permit fully closed processing and are disposable, providing compartmentalization that can greatly mitigate a contamination event. The control framework for successful implementation of SUBs includes addressing the critical nature of container closure integrity. In situ pressure-decay bag testing prior to use can detect leak sizes 300 micrometers (µm) and larger. Another critical concern for flexible containment systems is microbial ingress, which can occur with leaks as small as 10–22 µm. To prevent this, the SUB is maintained at positive pressure relative to the room.
Stainless steel bioreactors, equipment, and piping require changeover and cleaning to mitigate contamination. Process closure of the downstream manufacturing platform also increases operational flexibility, simplifies changeover, and reduces risk of virus contamination. Closure of existing stainless steel chromatography and tangential flow filtration skids by leveraging single-use interconnections creates a stainless steel–disposable hybrid solution, where many components are single-use gamma-irradiated tubing assemblies, filters, and bags.
Personnel, Materials, And Waste Flows
Personnel, materials, and waste flows are controlled via facility floor plans and room segregation. Access to GMP areas is limited to key staff and strictly monitored. GMP gowning and manufacturing operations, including manipulations, prevent introduction (or reintroduction) of viruses. All personnel entering processing areas must do so via designated gowning rooms and adhere to the requirements for that room. Persons with apparent illnesses are excluded from contact with raw materials, process intermediates, packaging materials, and the product until their conditions are corrected or they have been determined not to jeopardize product safety.
Facility Cleaning And Disinfection
Stainless steel equipment is cleaned between uses and tested at product changeover. Soil or contaminant-specific cleaning regimens must be verified or validated. All equipment and consumables introduced to the facility should be assessed for risk and exposed to disinfectants and cleaning agents that demonstrate the required removal and inactivation properties. In addition to other recommended contaminant-specific cleaning agents, 70% isopropyl alcohol—shown to have viricidal activity against retrovirus18 —is used routinely for hand spraying and equipment wipe-down. For part washing, a combination of caustic washing agents and high temperature provide effective inactivation. A strict rodent and insect control policy must also be in place.
“This Holistic Approach To Managing Virus And TSE Risk Provides Confidence That The Clinical Materials Supplied To Patients Are Free From The Risk Of Contamination”
Sodium hydroxide (NaOH) is used as a sanitizing solution for column resins.19 Even highly resistant non-enveloped viruses such as canine parvovirus and SV40§ are inactivated by NaOH, and enveloped viruses (such as influenza) are also effectively removed.
Virus Safety Triangle
Testing
Virus testing is a normal and routine part of biopharmaceutical manufacturing and quality control testing. According to ICH Q5A,1 appropriate testing for viruses must be carried out on the master cell bank (MCB), and must include both in vitro and in vivo tests. In vitro tests are carried out on a batch basis. More extensive testing is performed on cells at the limit of in vitro cell age, which should be evaluated for endogenous viruses that may have been undetected in the MCB and working cell bank. In vivo tests are conducted in rodents and embryonated eggs.
Detection
The rapidly expanding field of molecular virology and associated methodologies has developed extensive tools to analyze materials at all points of the manufacturing process for the presence of virus. These new methods, which include broad-range polymerase chain reaction as well as mass spectrometry, microarrays, and massively parallel sequencing (also referred to as next-generation sequencing), can detect a broad range of viruses both known and unknown.20 The advantages and disadvantages of these new methods and their ability to complement routine methods for virus detection form the basis of a continued dialogue between industry and regulatory agencies. Questions include whether nucleic acid detection is indicative of a complete genome or, more importantly, a live virus.
In cases of facility contamination there is no doubt that these methods offer a distinct advantage for rapid identification of the contaminant. Novel molecular methods also help reduce, refine, or replace live animals in biosafety testing. In addition, these new methods are increasingly being used to support the three-pronged approach to virus safety (i.e., the virus safety triangle), which has delivered safe products to patients over a considerable period of time.
Virus clearance validation
The third arm of the virus safety triangle is virus clearance validation (VCV). Typically carried out as part of the manufacturing process for clinical material, VCV requires a demonstration of clearance for two model viruses. Mechanisms of removal include partitioning or inactivation; the log reduction values are additive. For biologics license applications or marketing authorization application submissions, two additional model viruses are used. The aim is to obtain a clearance factor of < 1 virus-like particle in 106 doses of drug for both clinical and commercial material. A calculation is performed to determine the downstream load using the virus counts in the fermenter harvest. The amount of retrovirus present in fermenter harvests is measured ideally for three batches.
§ Simian vacuolating virus 40, or Simian virus 40, is an oncogenic polyomavirus that induces cancers in laboratory animals and may also affect humans.
- 18Corless I., A. Lisker, and R. W. Buckheit. “Decontamination of an HIV-Contaminated CPR Manikin.” American Journal of Public Health 82, no. 11 (1992): 1542–1543. http://ajph.aphapublications.org/doi/pdf/10.2105/AJPH.82.11.1542
- 19GE Healthcare. “Use of Sodium Hydroxide for Cleaning and Sanitization of Chromatography Media and Systems.” Application note 18-1124-57 AI. 14 November 2014. http://www.processdevelopmentforum.com/ppts/posters/NaOH_CIP_paper_18112457AI.pdf
- 1
- 20Parenteral Drug Association. “Emerging Methods for Virus Detection.” Technical Report No.71. 2015. www.pda.org
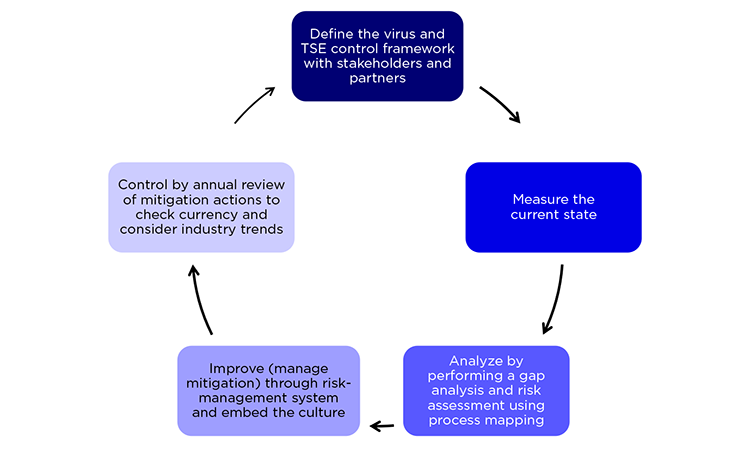
Risk-management Cycle
TSE and virus control requires a continuous improvement cycle. First, the current state is measured; process mapping analysis then identifies gaps and indicates necessary mitigations via a risk-management system. An annual review of mitigation actions to check currency and industry trends helps define best practices and provides continuous control (Figure 3).
Conclusions
This article outlines the risks that viruses and TSE agents pose to the manufacture of biopharmaceuticals and describes a holistic control strategy that promotes the provision of safe medicines to patients. Because these agents differ significantly in their physicochemical properties, they present unique challenges and require different approaches to risk mitigation.1 ,5 While raw material, component sourcing, vendor oversight, and certification controls are important, virus-risk reduction also relies on the application of both novel and routine methods of detection and virus clearance validation.
TSE control relies on documented risk assessments, including raw material sourcing, elimination of ADIs, and effective manufacturing controls such as alkaline hydrolysis and high heat extremes for product-contact materials, including tallow derivatives. While TSE clearance and detection methods are available, they are not routinely applied for biopharmaceutical manufacturing processes due to control of ruminant risk materials at source.
The control framework described here includes appropriate manufacturing facility design; HVAC system control; ISO room classification; architectural segregation; appropriate equipment types (stainless steel, single use, or hybrid); waste, materials, and personnel flows; cleaning and disinfection; pest control; and operator health policy. Internal and third-party control is governed by regulatory guidelines in addition to local policies and procedures monitored by quality assurance personnel.
Management of virus and TSE risk is a continuous process that involves gap analysis, improvement practices, and control measures. New control methods continue to be developed and implemented by the pharmaceutical industry. Gaps can be closed by adopting industry best practices and routine
oversight of the operations by quality assurance and compliance personnel. This holistic approach, using a three-pronged strategy for virus control and a TSE risk-assessment process, provides confidence that the materials supplied to patients are safe and free from virus and TSE contamination.


