Cleaning Validation Considerations for Automated Washing Systems

What is cleaning validation and where does it fall in the life cycle validation scheme? How can an automated washing system be validated? This article provides insights that may help answer these questions.

Analytical and sampling methods
As defined by the US Code of Federal Regulations, Title 21, Part 211, Subpart I, Section 211.165, “The accuracy, sensitivity, specificity, and reproducibility of test methods employed by the firm shall be established and documented.” 13
ICH Q2B guidance—a harmonized approach to the requirements for analytical method validation—provides additional information.14 ICH Q2B guidance was not developed specifically for analytical methods used in cleaning validation; the required elements of linearity, precision, range, robustness, accuracy, ruggedness, specificity, limit of quantitation, and limit of detection, however, are commonly applied to analytical method validations for cleaning validation (Table B).2
Analytical methods can fall into two categories:
Specific methods (preferred) identify the number of targeted species found in the presence of expected interferences. These methods can be ultra-performance liquid chromatography, ion chromatography, and atomic absorption.
Nonspecific methods, which can be total organic carbon (TOC), conductivity, and titration, take into account residue from all contributing factors. Visual inspection should be included for each item removed from the parts washer, and the procedure should classify a course of action for observed scratches, etching, dents, dings, and the like. These items may not be cleaning-related visual failures, but could be due to handling or wear.
The most common sampling methods are surface swabbing and rinse sampling. A less common procedure is direct surface sampling with an instrument such as a handheld Fourier transfer infrared spectroscopy or near-infrared spectroscopy.
For swab sampling, most companies use a single-swab method. In this approach, the surface is first sampled in overlapping strokes, then the swab is flipped over and used in overlapping strokes at a 90-degree angle to the first set (Figure 3). In the two-swab method, an area is sampled with a wet swab as noted above, then sampled by a second dry swab utilizing the same method. In both methods, water or another diluent is added to a vial with the swab or swabs. The analyte is extracted (or desorbed) from the swabs for analysis. Swab templates can be used for training, but not for actual part sampling, due to possible cross-contamination from the template to the swab.
In rinse sampling, small bottles, beakers, and other items are placed in a sterile plastic stomacher bag. Rinse water is added, the bag is sealed, and the contents mixed by shaking. A final rinse water sample or in-line measurement for conductivity and possibly TOC is used; the items must also be visually clean.
Whether using swab or rinse sampling methods, it is important to establish residue-recovery studies. The final rinse water specification and visually clean criteria should be confirmed with some level of surface sampling through swab, rinse, or direct methods.
Acceptance criteria
The cleaning validation master plan should help determine which residue to test for, and justify the limits established for surfaces or final rinse water samples. It is common to use purified water specifications for pH, conductivity, TOC, and microbial limits, along with a carryover estimate calculation based on residue toxicity. Residue limits are commonly calculated for drug active, cleaning agent, and bioburden, but may also include endotoxins, degradation products, excipients, or even the presence of color and fragrances. It’s helpful to include photographs or diagrams of the items to sample, and provide a rationale for selecting them. Such examples may be difficult-to-clean parts or those with the most complicated design.
In addition to setting limits on residue, it is often common to set acceptance criteria for the level of residual water left behind after the drying step. No droplets or residual water should remain on or in the items because this can lead to microbial growth.
If the cycle includes a sanitization/disinfection step, thermal strips or biological indicators can be used during the design phase to establish a log reduction. Chemicals, such as blends of hydrogen peroxide and peracetic acid (such as SporKlenz RTU disinfectant at a 1:50 dilution for 5 minutes), or hot water are effective sanitizers. Common time and temperature used for hot water pasteurization is 30 minutes at 63°C, 15 seconds for 72°C, and one second for 83°C.15
-

Figure 3: Swabbing method -

Figure 4: Semipermeable cover -

Figure 5: Riboflavin coverage testing
- 13US Code of Federal Regulations. Title 21, Part 211, Subpart I, Section 211.165: “Testing and Release for Distribution.” http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=211.165
- 14US Food and Drug Administration. Guidance for Industry. “Q2B Validation of Analytical Procedures: Methodology.” November 1996. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm073384.pdf
- 2
- 15Wardrip, C. L., J. E. Artwohl, and B. T. Bennett. “A Review of the Role of Temperature Versus Time in an Effective Cage Sanitization Program.” Contemporary Topics in Laboratory Animal Science 33, no. 5 (1994): 66-68 1994.
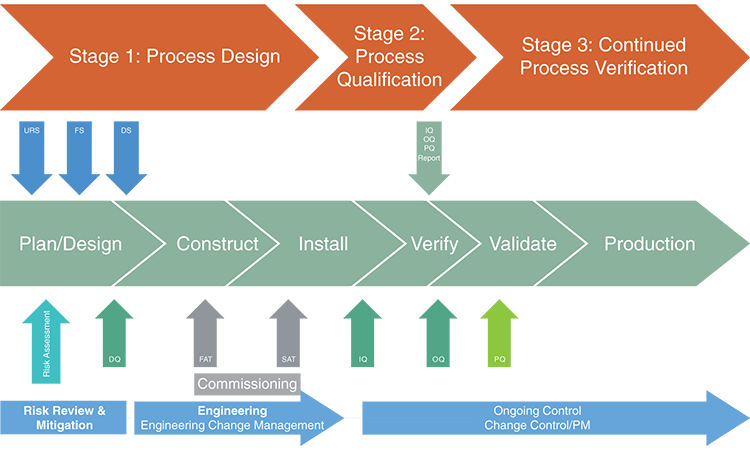
STAGE 2: PROCESS QUALIFICATION
Stage 2, qualification of the automated parts washer and cleaning validation could be approached as a readiness check. Before starting the process, the following should be confirmed:
- Cleaning documentation including protocols and operating procedures have been approved
- Personnel have been trained on the documentation and procedures
- Utility supply systems have been qualified
- Analytical methods and sampling procedures have been validated
- Suppliers of cleaning agents have been approved
- Automated washer equipment is fully functional
Qualification
Stage 2 typically includes installation qualification (IQ) and operation qualification (OQ) to determine that the automated washer:
- Has been installed as specified and the utilities are sufficient to maintain operation
- Is operating as specified
These procedures may include a repeat of the riboflavin coverage testing, a successful run of a complete cleaning wash cycle, verification that all alarms are functioning properly, and confirmation that sensors/probes are calibrated and functioning as designed.
The next step is to execute the performance qualification (PQ) of the washer. Sampling should be performed on the soiled parts to establish a baseline, and on the cleaned items to demonstrate that the final rinse water acceptance criteria corresponds to the cleanliness of the parts washed.
Cleaning validation
As noted above, the traditional cleaning validation (PQ) approach of evaluating three runs may not be applicable. Instead, the number of runs may depend on the testing performed during the Stage 1 design and risk assessment. Evaluating worst-case critical parameters is also not applicable because critical parameters identified during the design stage were identified and monitored or controlled. The goal of the PQ is to demonstrate that the normal operating cleaning cycle using the automated parts washer successfully removes the residue(s) of interest to predetermined acceptable limits.
The PQ process should be thoroughly documented and approved. Any deviations, changes, or OOS events should be recorded and a risk assessment performed to assess impact to the PQ activities.
| Description | Quantity | Height, mm | Outer diameter (OD) |
Weight, kg | Critical information |
Drawing or picture number |
Notes |
|---|---|---|---|---|---|---|---|
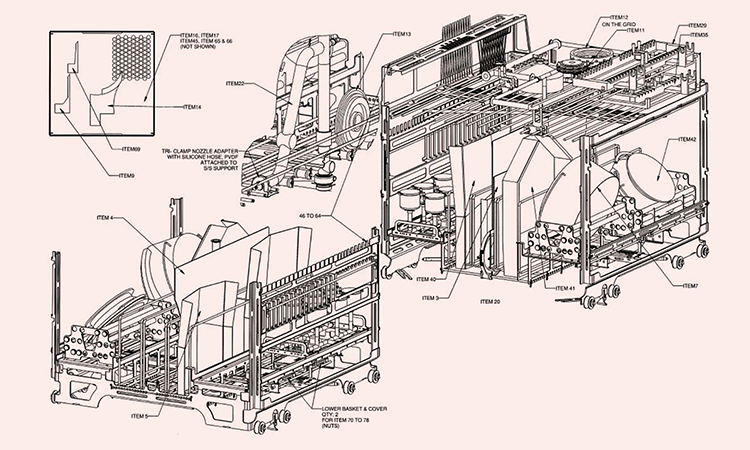
| Filling needle | 8 | 110 | 15 | NA | Process soil: low concentration protein |
 |
Photo 28 |
| Filling pump | 8 | 174,5 for pump 150 for plunger |
Pump OD 70,6 Plunger inner dia. 18 |
NA | Process soil: Low concentration protein, material: external is 316L SS, pump internal is porcelaine, can separate wash |
 |
Photo 29 |
| Glass bottle | 1 | 300 | 180 | NA | Process soil: low concentration protein |
 |
Photo 30 |
| Characteristic | Recommended criteria |
|---|---|
| Precision | Precision should be assessed using at least nine determinations (3 concentrations with 3 replicates each) covering the specified range and reported as Relative Standard Deviation (RSD). |
| Limit of quantitation (LOQ) | The LOQ can be estimated by measuring the baseline noise multiplied by 10. This value must be less than the cleaning validation acceptance limit. |
| Limit of detection (LOD) | The LOD can be estimated by measuring the baseline noise multiplied by 3. This value must be less than the cleaning validation acceptance limit. |
| Accuracy | Accuracy should be assessed using at least nine determinations (3 concentrations with 3 replicates each) covering the specified range and reported as percent recovery. The percent recovery should be close to 100%. |
| Specificity | Specificity may be demonstrated by comparing the test results of samples containing analyte plus other expected components versus samples of analyte only. |
| Linearity | Linearity should be established with a minimum of five concentrations and three replicates each. The coefficient of determination (R2) of the linear regression should be not less than 0.99. |
| Project scope | Available utilities | Washer location environment | |
|---|---|---|---|
|
|
|
|
| Loading accessories (racks) | |||
|
|||
| Standards and guidelines | |||
|
|||
| Design requirements | Documentation requirements | ||
| Electrical Instrumentation (preferred vendors, accuracy) Tagging for instrumentation Labeling |
Paper or Electronic Language |
||
Mechanical
|
Design documentation
|
||
Control system
|
Manufacturing documentation
|
||
Process monitoring
|
Process records
|
Manuals
|
Qualification
|
Stage 3: Continued Process Verification
The main purpose of the third life cycle stage is to provide continued assurance that the cleaning procedure is performing as expected, and that it remains in a state of control for the life of the product(s) being manufactured. In this stage, the facility is manufacturing product and the cleaning procedure and automated washer are operating within the normal range.
Stage 3 typically includes regular reviews of the cleaning performance, cleaning procedures, training program, change controls, deviations, corrective and preventive actions, and preventive maintenance activities.
Preventive maintenance
The initial preventive maintenance program of the automated washer and parts should be based on the manufacturer’s recommendations, and adjusted as the equipment ages or real-time performance metrics support indicate. Laboratory testing can also be used to investigate items such as compatibility between gasket and tubing materials.
| Changes to | May affect |
|---|---|
| Detergent | Cleanability of the soils |
| Cleaning parameters | Cleanability of the soils |
| Analytical method | Surface coverage, equipment drainability, change over time |
| Personnel | Training and level of experience |
| Dirty hold time | Cleanability of soils, level of bioburden |
| Cleaning hold time | Extraneous matter, bioburden |
Stage 3 includes trend analyses of the measured CPPs and CQAs (e.g., online conductivity and TOC of the final rinse water) as well as drying temperature/time and ramp rates, which can increase cycle times.18 ,19 Data trending helps supports corrective actions prior to deviations or OOS results, which can compromise the quality of products manufactured.
The life cycle approach, which emphasizes understanding and effective continuous verification of the cleaning process, should be open to change control to improve its efficiency and drive down production costs while maintaining high quality standards. Table D lists changes to the cleaning process and possible results of the of the change.2
Conclusion
The cleaning life cycle approach (design, qualification, and continued verification) focuses on design and monitoring of the cleaning process as well as a better understanding of the design process (critical parameters and URS of the automated parts washer). This promotes continuous improvements and real-time science-based responses to OOS results and change management. Industry tools are the backbone to the life cycle approach and these elements can be incorporated into cleaning validation when using automated parts washers.
Whether using swab or rinse sampling methods, it is important to establish residue-recovery studies.
- 18Dion, M., O. Van Houtte, and G. Verghese. “On-Line TOC Monitoring in GMP Parts Washers.” Pharmaceutical Engineering 34, no. 2 (March/April 2014): 80–87.
- 19Bader, K., J. Hyde, P. Watler, and A. Lane. “On-Line Total Organic Carbon (TOC) as a Process Analytical Technology for Cleaning Validation Risk Management.” Pharmaceutical Engineering 29, no. 1 (January/February 2008): 8–20.
- 2


