Improving Study Start-Up SSU & the Clinical Trail Continuum

Workflow-based technology in the clinical trial continuum encourages process optimization, helps break down silos, enhances performance quality, and has a measurable effect on the electronic trial master file.
The focus on technology as a driver of performance improvement in clinical trials is intense, but despite years of valiant efforts, study execution remains far from optimal. For study start-up (SSU), one of the most complex parts of clinical trials, the data are dismal: Contract cycle times have doubled from an industry median of 1.5 months in 2009–2011 to more than three months in 2014–2015.1 Nearly 50% of clinical trials are behind schedule, with slow patient enrollment cited as the top reason.2 Research also suggests that for Phase II and III trials, a lengthy 16.7 months is typically required to initiate all approved investigative sites.3
These statistics are not surprising, given the findings of a new SSU process survey conducted by the Tufts Center for the Study of Drug Development (CSDD), in which 35% of respondents still rely on spreadsheets to launch trials, 2% use paper-based systems, and 19% were unsure, as this function is outsourced to a contract research organization (CRO).4
While technology remains critical, as emphasis shifts to process optimization it may be only part of the solution. Since the introduction of electronic data capture in the 1990s multiple providers have entered the marketplace, offering point solutions∗ to improve quality in clinical trials. Two decades later, however, stakeholders have learned that point solutions can hinder the flow of data across the continuum,5 causing already entrenched silos to dig in further. The need to move to the bigger picture—the overall process—should resonate with stakeholders responsible for SSU management.
Although there is no industry-wide definition of SSU, a series of steps—from site identification to contract and budget negotiations to site initiation—are generally associated with starting a study. To improve performance quality for these elements, two factors are needed: an end-to-end solution and support from top management, i.e., the chief executive officer, chief information officer, chief medical officer, and others who comprise the so-called C-suite.
An end-to-end solution with workflows that aggregate data from disparate sources can draft documents in the correct format from the start and release them downstream into the trial master file (TMF) or electronic trial master file (eTMF). This approach can break down silos that have long performed in isolation with little understanding of what the next department needs to fulfill its regulatory obligations and achieve targets measured by performance metrics.
As for the C-suite, the importance of buy-in from upper management cannot be overstated. Such input provides the critical impetus and strategic insight to align with the sponsor’s goal for developing better therapies more quickly. Without management direction, efforts to jump-start overall performance optimization tend to flounder as departments retreat to their silos.
In short, tools are essential, but they don’t create a master craftsman. Real expertise comes from combining experience with the authority and talent to influence the way studies are conducted from an operational perspective. Research suggests that organizational issues become strategic and of interest to upper management once they have proven relevance to performance.6
This article focuses on how workflow-based technology encourages process optimization and how these improvements enhance performance quality. Purpose-built SSU solutions can identify the documents needed to conform to downstream regulatory requirements, and can also signal bottlenecks or breakdowns in study execution. This approach helps to avoid rework, delays, and cost overruns; improves cycle times; and facilitates audit readiness.
Where’s your focus?
Transforming these clinical trial elements17 can lead to quality improvements:
- Contractual agreements between sponsors, institutions, and investigators
- Investigator recruitment
- Participant recruitment plan
- Quality control systems
- Data collection, management, and analysis
- Data standards
- Regulatory approval to conduct a clinical trial (e.g., IND in the United States)
- Coordination of global investigators and research sites
∗ According to PC Magazine Encyclopedia, point solutions “[solve] one problem without regard to related issues. Point solutions are widely used to fix a problem or implement a new service quickly.”
Improving The Process
The need to improve the clinical trial process, starting with the SSU, is a long-standing industry battle cry. A seminal 2012 report from the Institute of Medicine (IOM) confronted this issue head-on by encouraging transformation of the clinical trials enterprise through quality improvement efforts.7 ,8 The report contains a lengthy discussion on infrastructure improvements, identifies key elements, and recommends the following:
- Use “creative destruction” to replace old clinical trial business models with newer forms that complement advances in technology.7
- Trade outmoded mechanisms for newer technologies such as web-based clinical trials and smartphones.8
- Engage in more strategic planning and consider new organizational structures for entities that conduct clinical trials.8
Since the IOM report was published, process improvement has emerged as a hot-button issue, as evidenced by the expanding volume of literature on the subject. Some articles, for example, confirm the widely acknowledged challenges linked to contract and budget negotiations. Martinez et al. found those tasks to be the most time-consuming part of the study activation process and to be widely variable due to lack of standardized processes.9 Using a simulation model, they determined that increasing the efficiency of contract and budget development would reduce activation time by 28%.
Other articles describe the need for an organized Six Sigma approach to improve SSU processes, in which steps are carefully defined and continuous improvement becomes standard practice.10 For SSU, those steps involve accessing process, people, data, and systems for activities that range from site selection to site activation.
More recently, the survey of 591 clinical trial stakeholders conducted by the Tufts CSDD in Q1 2017 determined that a mere 8% of sponsors and 14% of CROs are extremely satisfied with their SSU processes. By comparison, approximately 40% are either somewhat or completely unsatisfied with those processes.4 Not surprisingly, respondents reporting that they are extremely satisfied have cycle times 57.5% shorter than those that claim to be completely unsatisfied.
Better Quality Fewer Silos
While the industry tries to implement processes that improve SSU quality, regulatory efforts may be the driving force. The November 2016 release of the first new good clinical practice (GCP) guideline in 20 years was a major step forward.11 Put forth by the International Conference on Harmonization (ICH), the guideline, known as ICH-GCP E6(R2), is an addendum to the original statement from 1996. It includes a new section focused exclusively on risk-based quality management. It states that the sponsor should implement a system to manage quality from the start, and throughout all stages of the trial process. The new section also addresses topics such as critical process and data identification followed by subsections focused on risk factors, namely risk identification, risk evaluation, and risk control.
The guideline acknowledges that technology has advanced to the point that it can support processes and generate data that provide actionable insights into risks and study bottlenecks. As described in the new guideline:
Evolutions in technology and risk management processes offer new opportunities to increase efficiency and focus on relevant activities. When the original ICH E6(R1) text was prepared, clinical trials were performed in a largely paper-based process. Advances in use of electronic data recording and reporting facilitate implementation of other approaches. . . . Therefore, this guideline has been amended to encourage implementation of improved and more efficient approaches to clinical trial design, conduct, oversight, recording and reporting while continuing to ensure human subject protection and reliability of trial results.11
| Sites | Very or somewhat shorter, % | Very or somewhat longer, % | |
|---|---|---|---|
| Sponsors | Repeat | 18.9 | 27.5 |
| New | 15.9 | 35.0 | |
| CROs | Repeat | 36.1 | 15.3 |
| New | 23.9 | 15.5 | |
| Centralized SSU | Repeat | 35.9 | 17.1 |
| New | 27.9 | 20.7 | |
| Decentralized SSU | Repeat | 17.7 | 27.9 |
| New | 13.5 | 33.3 | |
| Source: Tufts Center for the Study of Drug Development, 2017. Used with permission. | |||
Application program interfaces (APIs) are an example of technology that has greatly increased clinical trial efficiency. These tools integrate cloud-based eClinical solutions such as electronic data capture, the investigator portal, the data warehouse, and the clinical trial management system. APIs allow sponsors and CROs to link stakeholder data across the globe, irrespective of the systems they are using, optimizing the data flow across the clinical trial continuum and eventually releasing it into the eTMF.</p> <p>Unfortunately, entrenched silos such as site identification, clinical development, data management, contracting, and regulatory affairs have long stymied these data flow efforts because they often have minimal understanding of what is needed downstream. This approach is often dubbed the “throw it over the wall” mentality, meaning that once a specific department has completed its work, it is tossed to the next department in assembly-line fashion.<fn value=">Doyle J., D. DeBard, A. Butcher, et al. “Partnerships 2010: Let’s Make a Deal: Breaking Out of Organizational Silos—Bridging the Gap from Clinical Development to Commercialization.” KNECT 365 Clinical Trials.
This awkward management style is a root cause of problems with the TMF and eTMF. Information about the standardized taxonomy and metadata provided in the TMF reference model is not typically shared with SSU team members, so they are frequently unaware of which documents are needed or the required format for release into the eTMF. Since start-up generates almost half of the artifacts found in the TMF—data files, documents, digitized content, and media—this can create challenges for the regulatory group tasked with mapping documents to the TMF and indexing the metadata.13
“Workflow-based technology in the clinical trial continuum encourages process optimization, helps break down silos, enhances performance quality, and has a measurable effect on the ETMF”
Fortunately, technology provides the opportunity to rethink the inefficiencies of silos. Some stakeholders want to move away from vertical silos and “think horizontally.” This method uses automation and workflows to integrate operational data across all functions, making it easier to extract meaningful insights from those data.14 Some also believe that bringing interdependent functions together using technology and critical teams will help navigate the highly complicated global regulatory maze.15
EMBRACING WORKFLOWS
Optimizing study conduct starts with embracing a workflow-based process that defines the documents needed for the SSU. This method boosts quality by preparing documents that are accurate, complete, and in alignment with the eTMF format established by a sponsor’s or CRO’s regulatory team, enhancing audit readiness.
The 2012 Tufts CSDD study highlights sponsors’ and CROs’ strong need for improvement.4 Both had lengthy site start-up cycle times—in the range of 4 months for repeat investigative site CROs and 5 months for sponsors; times were even longer for new sites. The study also revealed that site startup process cycle times were substantially longer than they were just 3 years earlier, particularly for sponsors (Table A). Companies with centralized SSU functions did, however, see strong improvement.
Given these statistics, a workflow-based platform that facilitates quality efforts is a sensible option. The tool’s integrated data from several e-clinical solutions provides an end-to-end continuum that allows properly formatted documents and structured artifacts to flow into the eTMF. With the help of this tool, moreover, documents eventually needed for the eTMF can be defined up front, during SSU. This is a major advantage because within those documents more than 400 draft and supporting artifacts can be structured, resulting in a final set of approximately 60 artifacts that will be released into the eTMF.
CASE STUDY
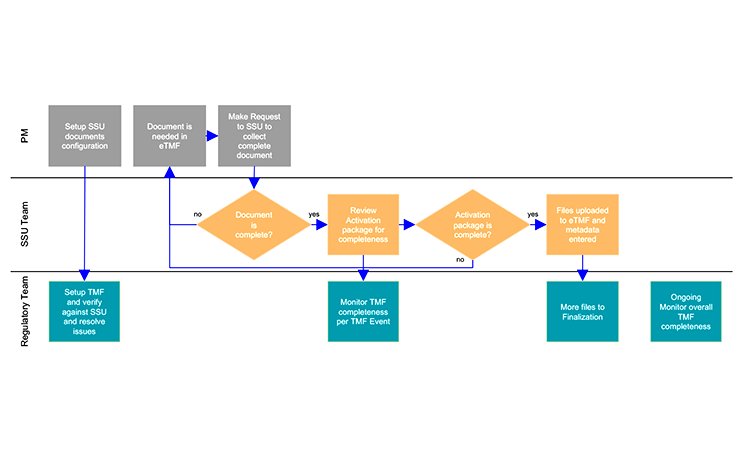
A recent case study details how a workflow-based tool helped a large CRO improve eTMF quality. The company had a history of uploading site documents and associated metadata into the eTMF manually.16 With the complexities of SSU and global regulatory requirements, determining when all necessary documents appeared in the eTMF required multiple quality control checks, which proved a tremendous drain on resources. In addition, as the CRO scaled clinical trials, the efficiency and quality of data entering the eTMF created audit risk and increased costs for full-time equivalents.
To manage this problem, the CRO implemented a tool that guides SSU workflows, documents, and submission collections with mappings to the TMF reference model. Using an API, artifacts and metadata were delivered to the eTMF only when specific business conditions or events occurred. As a result, the CRO was able to handle an estimated 20% increase in studies with the same staff levels, while also reducing the number of rejections per study site for artifacts where either the wrong version had been used or associated metadata were missing. Figure 1 depicts this workflow.
- 1KMR Group. “Site Contracts from Weeks to Months: Results from KMR Group’s Site Contracts Study.” Press release. August 24, 2016. http://www.biospace.com/News/site-contracts-fromweeks-to-months-results-from/430240
- 2Gwinn B. “Metrics for Faster Clinical Trials.” PharmaVoice, June 2011. http://www.pharmavoice.com/article/2011-06-metrics-for-faster-clinical-trials
- 3goBalto and Tufts Center for the Study of Drug Development. “‘START’ (Start-up Time and Readiness Tracking) Study.” https://www.gobalto.com/research-report-tufts-2012
- 4 a b goBalto. START II (Start-up Time and Readiness Tracking) Study. 2017.
- 5Goldsmith J. “It’s Time to Transform Clinical Trials Operations.” Applied Clinical Trials, 1 August 2016. http://www.appliedclinicaltrialsonline.com/it-s-time-transform-clinical-trial-operations
- 6Dutton J. E., and S. J. Ashford. “Selling issues to top management.” Academy of Management Review 18, no. 3 (July 1993): 397–428. http://webuser.bus.umich.edu/janedut/Issue%20Selling/sellingissues.pdf
- 17Eisenberg, Paul, Petra Kaufmann, and Janet Woodcock. “Developing a Clinical Trials Infrastructure.” Appendix G in Envisioning a Transformed Clinical Trials Enterprise in the United States: Establishing an Agenda for 2020: Workshop Summary. Institute of Medicine, 2012. https://www.ncbi.nlm.nih.gov/books/NBK114674
- 7 a b National Institutes of Health. Institute of Medicine. Envisioning a Transformed Clinical Trials Enterprise in the United States: Establishing an Agenda for 2020: Workshop Summary. 2012. http://www.fdanews.com/ext/resources/fi les/archives/4/4-17-12-trials.pdf
- 8 a b c Kramer J. M., and K. A. Schulman. “Transforming the Economics of Clinical Trials.” Appendix F in Envisioning a Transformed Clinical Trials Enterprise in the United States: Workshop Summary. 2012. https://www.ncbi.nlm.nih.gov/books/NBK114653
- 9 Martinez D. A., A. Tsalatsanis, A. Yalcin, et al. “Activating Clinical Trials: A Process Improvement Approach.” BioMed Central Trials 17, no. 106 (2016). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4765218/pdf/13063_2016_Article_1227.pdf
- 10goBalto. “Study Start-Up Leader or Laggard? Time to Assess Your Level of Maturity.” White paper. 2016. https://www.gobalto.com/whitepaper_study_startup_leader_or_laggard?hsCta-Tracking=19b10629-077b-41f5-9f31-e214502074d1%7Cadfa9a68-c6f4-4e2a-8122-2d545dbc2d88
- 11 a b International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Guideline. “Integrated Addendum to ICH E6(R1): Guideline for Good Clinical Practice E6(R2).” 9 November 2016. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R2__Step_4.pdf
- 13Trial Master File Reference Model. “Version 3.0 of the Trail Master File Is Here!” 16 June 2015. https://tmfrefmodel.com/2015/06/16/version-3-released
- 14Fassbender M. “The Rise of ‘Technology-Enabled’ Clinical Research Companies. Outsourcing-pharma.com. 17 January 2017. http://www.outsourcingpharma.com/Clinical-Development/The-rise-of-technology-enabled-clinical-research-companies
- 15Pisani J., and M. Lee. “A Critical Makeover for Pharmaceutical Companies. Overcoming Industry Obstacles with a Cross-Functional Strategy.” Strategy&. 10 January 2017. https://www.strategyand.pwc.com/reports/critical-makeover-pharmaceutical-companies
- 16goBalto. “Automating IP: Green Light Release to eTMF: Intelligent Submissions of eTMF Filings. Case study. 2016. https://www.gobalto.com/hubfs/docs/ETMF_CS_2SHEET-10NOV.pdf
PLAN EARLY AND BREAK DOWN SILOS
As clinical trial stakeholders ramp up eff orts to optimize the SSU process and begin the arduous task of dismantling silos, there is a growing recognition that technology is a critical component. Without it, sponsors and CROs will continue to experience the measurable ramifications of poor quality: delays, cost overruns, poor communication, and lack of audit preparedness. These problems can be avoided with the expanded use of workflow-based tools and performance metrics.
Such initiatives to improve quality are in early stages, but with the availability of solutions, transformational process changes can finally begin to happen. Planning up front for correctly formatted documents, artifacts, and associated metadata that will eventually be released into the eTMF will bring big changes to the typically inefficient chain of study execution. This is reflected in statistics that document just how intractable study start-up problems have become. Despite the presence of many point solutions, for example, 8 months remains the average time frame for moving from previsit to site initiation.3
Significantly, with the use of integrated information from disparate data sources, issues will be identified early on, rather than waiting until they reach the eTMF. This is because regulatory metrics derived from documents arriving in the eTMF are developed too late to provide proactive insight. But with a real-time workflow tool, insight from performance metrics can offer the transparency needed to take action in real time. By embracing this approach, complemented by support from key decision makers, it is possible to move the needle on process change and increase the likelihood of more predictable cycle times, better adherence to study budgets, and audit readiness.


