Falsified Medicines Directive 2011/62/EU was published by the European Parliament on 08-Jun-2011. The Directive was implemented to increase the security of the manufacturing and delivery of medicines across Europe, protect patients and prevent falsified medicines from entering the supply chain. Commission Delegated Regulation (EU) 2016/161 details the characteristics of the safety features, how medicine authenticity should be verified and by whom. The delegated Regulation, and the new medicine verification system it details, became effective on 09-Feb-2019. Regulation 2016/161 article 16.2, 23 and 25 pertain to the verification, decommissioning and re-commissioning of commercial serialized packs. There is no current industry standard to determine how this regulation impacts the packaging, labelling and distribution (PLD) of commercial products used in clinical supply chains and who is responsible as manufacturer at multiple processing stages in the supply chain. ISPE task team is aiming to understand and share experiences from industry post implementation of the regulation and to seek continuous feedback with the long term aim of aligning approaches within the IMP community of practice.
According to Art. 16.2, manufacturers have to execute two key operations on the commercial packs prior to repackaging or relabeling them for use in CTs:
- Verify integrity of the tamper evident feature
- Decommission Unique Identifiers in the National Medicine Verification systems
Commission Q&A #1.6 indicates FMD requirements “apply up to the moment it becomes known which batch/unit will be used for research and development trials”. It is not clear that interpretation is consistent across member states.
Art. 16.2 addresses responsibilities of manufacturers holding both commercial and IMP Manufacturing Authorizations, however there is no explicit information about when commercial material entering the clinical supply chain, for use as IMP, has to be decommissioned and kit integrity verified or by whom this activity should be completed, considering the complexity and variability of clinical supply chains.
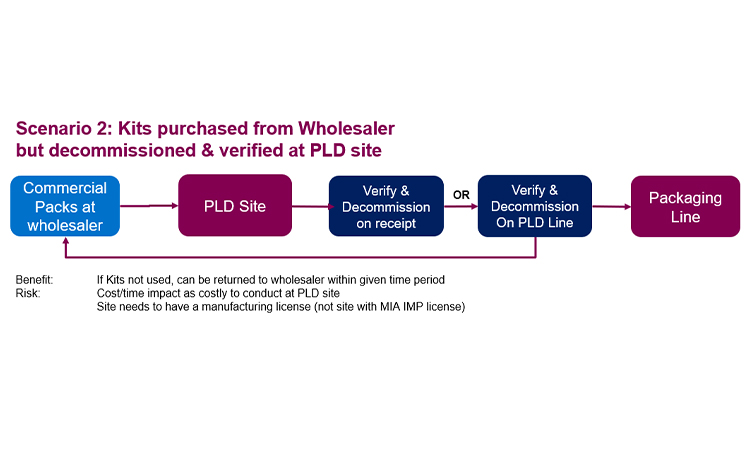
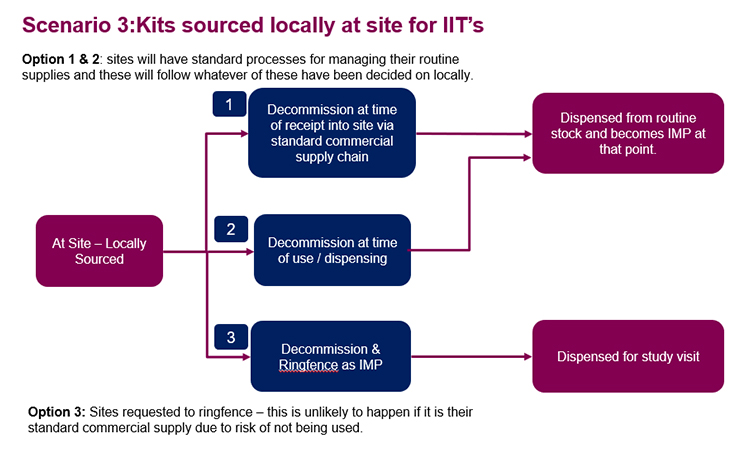
Depending on the supply chain configuration, different scenarios could be considered relative to product decommissioning. As the majority of products intended for use in CTs are sourced by the sponsor through a supplier (wholesaler / manufacturer) for repackaging and relabeling as IMP, this will be considered in detail below. This configuration offers three scenarios:
- It is known at the time of product purchase that it will be used in CTs. Suppliers could execute the decommissioning prior to material shipment to the sponsor or its subcontractor. This scenario reduces downstream impact.